- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(1); 2025 > Review
-
Minireview
Advances in functional analysis of the microbiome: Integrating metabolic modeling, metabolite prediction, and pathway inference with Next-Generation Sequencing data -
Sungwon Jung1,2

-
Journal of Microbiology 2025;63(1):e.2411006.
DOI: https://doi.org/10.71150/jm.2411006
Published online: January 24, 2025
1Department of Genome Medicine and Science, Gachon University College of Medicine, Incheon 21565, Republic of Korea
2Gachon Institute of Genome Medicine and Science, Gachon University Gil Medical Center, Incheon 21565, Republic of Korea
- Sungwon Jung E-mail: sjung@gachon.ac.kr
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- This review explores current advancements in microbiome functional analysis enabled by next-generation sequencing technologies, which have transformed our understanding of microbial communities from mere taxonomic composition to their functional potential. We examine approaches that move beyond species identification to characterize microbial activities, interactions, and their roles in host health and disease. Genome-scale metabolic models allow for in-depth simulations of metabolic networks, enabling researchers to predict microbial metabolism, growth, and interspecies interactions in diverse environments. Additionally, computational methods for predicting metabolite profiles offer indirect insights into microbial metabolic outputs, which is crucial for identifying biomarkers and potential therapeutic targets. Functional pathway analysis tools further reveal microbial contributions to metabolic pathways, highlighting alterations in response to environmental changes and disease states. Together, these methods offer a powerful framework for understanding the complex metabolic interactions within microbial communities and their impact on host physiology. While significant progress has been made, challenges remain in the accuracy of predictive models and the completeness of reference databases, which limit the applicability of these methods in under-characterized ecosystems. The integration of these computational tools with multi-omic data holds promise for personalized approaches in precision medicine, allowing for targeted interventions that modulate the microbiome to improve health outcomes. This review highlights recent advances in microbiome functional analysis, providing a roadmap for future research and translational applications in human health and environmental microbiology.
Introduction
Glossary of Technical Terms
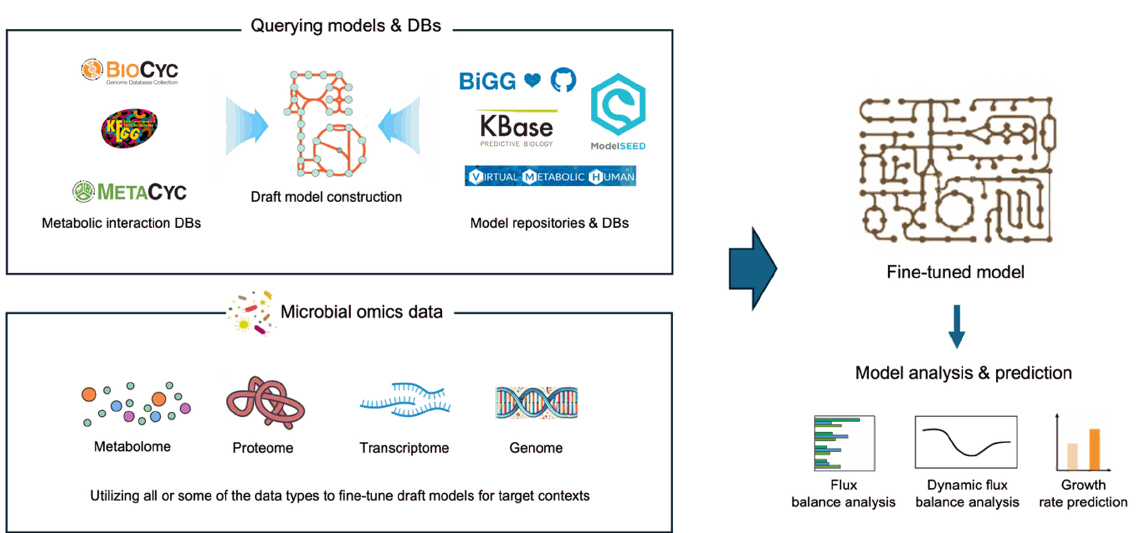
GEMs for Microbiome Research
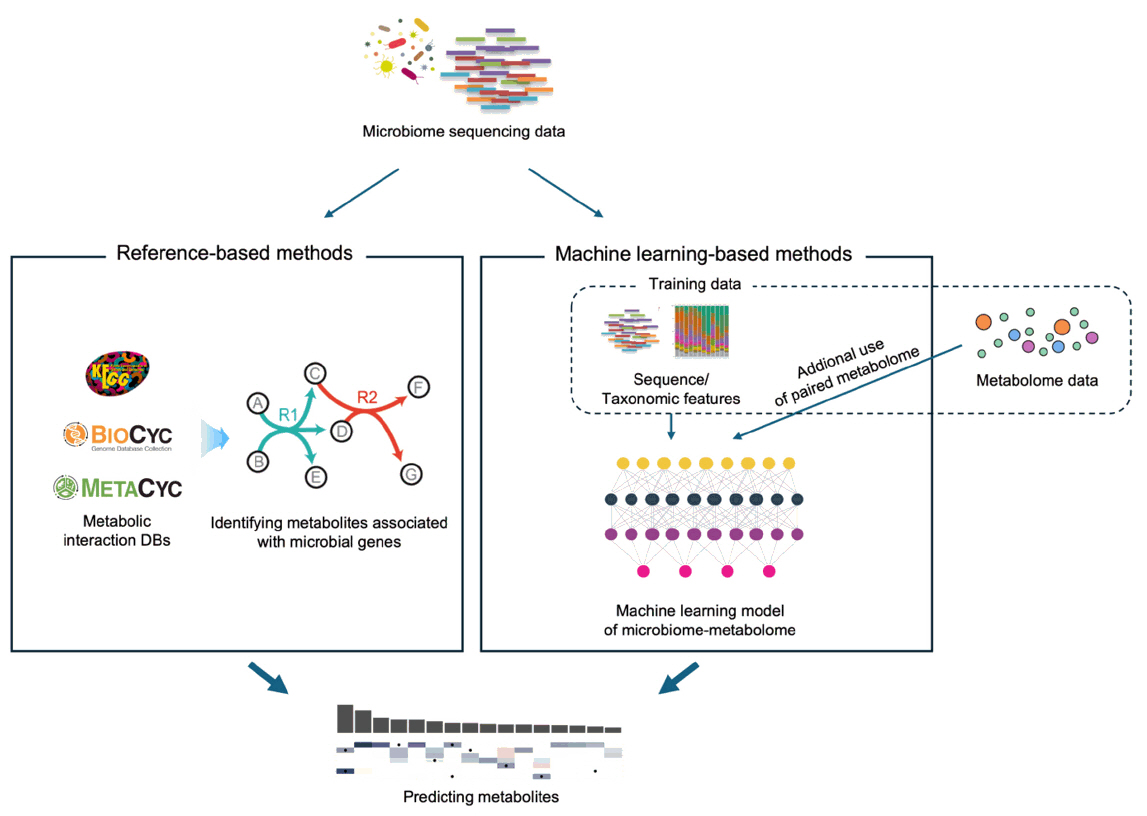
Predicting Metabolite Profiles from Microbiome Sequencing Data
Functional Pathway Analysis Tools for Microbiome Data
Limitations and Challenges of Current Approaches
Conclusion
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government Ministry of Science and ICT (MSIT) [NRF-2022R1A2C1007345].
Conflict of Interest
The author declares that there is no competing interest.
| Name | Purpose/Functionality | Key features |
|---|---|---|
| AGORA2 (Heinken et al., 2023) | Personalized and predictive modeling | Models of 7,302 microbial strains |
| Model repository | Information on 98 drugs and relevant enzymes | |
| BacArena (Bauer et al., 2017) | Individual-based metabolic modeling of microbial communities | Integrates FBA with individual-based modeling |
| Modeling spatial and temporal dynamics | ||
| BiGG (King et al., 2016) | Repository for GEMs | 77 manually curated GEMs |
| Knowledge integration | Supporting various model formats | |
| Community collaboration | Supporting web API | |
| CarveMe (Machado et al., 2018) | Fast reconstruction of GEMs for microbial species and communities | Top-down approach using a universal model for scalable model generation |
| Automated gap-filling for improved growth phenotype predictions | ||
| COBRA (Heirendt et al., 2019) | Constraint-based modeling of biochemical networks | Extensive support for FBA and omics data integration |
| High-performance solvers for multi-scale and genome-scale models | ||
| COMETS (Dukovski et al., 2021) | Dynamic simulation of microbial community interactions | Spatially structured dFBA |
| Supports Python and MATLAB interfaces for customized simulations | ||
| DyMMM (Zhuang et al., 2011) | Simulating interactions and competition in microbial communities under dynamic conditions | Integrates genome-scale models for multi-species interactions |
| Predicts community dynamics under varying environmental conditions | ||
| jQMM (Birkel et al., 2017) | Modeling microbial metabolism and analyzing omics data | Combines FBA and 13C metabolic flux analysis |
| Uses 13C labeling data for genome-scale model constraints | ||
| KBase (Arkin et al., 2018) | Data sharing, integration, and analysis for systems biology | Diverse data integration (genomes, biochemistry) |
| Web-based interface with data provenance | ||
| MCM (Louca & Doebeli, 2015) | Modeling multi-species microbial communities with genome-based metabolic models | Statistical parameter calibration with experimental data |
| dFBA for metabolic interaction simulation | ||
| metaGEM (Zorrilla et al., 2021) | Reconstruction of GEMs from metagenome | End-to-end pipeline for community-level metabolic interaction simulations |
| Generates personalized metabolic models from metagenome-assembled genomes (MAGs) | ||
| MetExplore (Cottret et al., 2018) | Collaborative curation and exploration of metabolic networks | Data mapping for multi-omics integration |
| Sub-network extraction and interactive visualization | ||
| Microbiome Modeling Toolbox (Heinken & Thiele, 2022) | Efficient modeling and analysis of microbiome communities | Parallelized generation of personalized microbiome models |
| Visualization and statistical analysis for model comparison | ||
| MMinte (Mendes-Soares et al., 2016) | Predicts metabolic interactions among microbial species in a community | Pairwise interaction analysis under different metabolic conditions |
| Modular interface with independent functionalities for flexibility | ||
| ModelSEED (Henry et al., 2010) | High-throughput generation and optimization of GEMs | Automated reconstruction pipeline from genome annotation to draft models |
| Integrates gap-filling for biomass production and growth simulation | ||
| OptCom (Zomorrodi & Maranas, 2012) | Multi-level optimization for modeling metabolic interactions in microbial communities | Balances individual VS. community fitness criteria |
| Captures various interaction types (positive, negative) for multiple species | ||
| RAVEN (Wang et al., 2018) | Reconstruction and analysis of GEMs | Supports de novo model reconstruction using KEGG and MetaCyc databases |
| Integration with COBRA Toolbox for compatibility and bi-directional model conversion | ||
| SteadyCom (Chan et al., 2017) | Predicting microbial community composition and maintaining steady-state growth | Ensures constant community growth rate across all species |
| Supports flux variability analysis to explore metabolic flexibility | ||
| VMH (Noronha et al., 2019) | Integration of models with extrinsic factors such as nutrition and disease | Extensive data coverage (Recon3D human model, 818 microbial models, disease/nutrition information) |
| Method type | Name | Data requirements | Advantages | Limitations |
|---|---|---|---|---|
| ML-based | LOCATE (Shtossel et al., 2024) | Paired microbiome (16S or metagenomics) and metabolomics data | Latent representation and low data requirement for training | Limited cross-dataset generalization |
| Reference-based | Mangosteen (Yin et al., 2020) | Microbiome sequencing data | Utilizes curated databases | Limited by database coverage |
| ML-based | MelonnPan (Mallick et al., 2019) | Amplicon or metagenomic sequencing data, paired with metabolomic data for training | Predicts metabolomic profiles from metagenomic data | Requires training data and limited generalization |
| ML-based | MiMeNet (Reiman et al., 2021) | Paired microbiome (metagenomic taxonomic/functional) and metabolome data | Improves prediction via multivariate learning | Performance depends on dataset size |
| Reference-based | MIMOSA2 (Noecker et al., 2022) | Paired microbiome (16S or metagenomics) and metabolomics data | Infers mechanistic microbe-metabolite links | Limited to environments represented in reference databases |
| Name | Approach | Input data | Unique features |
|---|---|---|---|
| bioBakery (Beghini et al., 2021) | Reference-based, assembly-independent profiling | Metagenomic and metatranscriptomic sequences | Integrates taxonomic, strain-level, functional, and phylogenetic profiling |
| METABOLIC (Zhou et al., 2022) | High-throughput metabolic and biogeochemical profiling | Genomes from isolates, metagenome-assembled genomes, or single-cell genomes | Community-scale functional networks |
| MintTea (Muller et al., 2024) | Identification of multi-omic modules | Taxonomic, Functional, Metabolome profiles | Integration of multi-modal data and identifying predictive modules |
| PICRUSt2 (Douglas et al., 2020) | Phylogenetic placement and hidden state prediction | 16S rRNA gene sequences | ASV compatibility, supports custom databases |
- Altman T, Travers M, Kothari A, Caspi R, Karp PD. 2013. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinform. 14: 112.ArticlePDF
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, et al. 2018. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat Biotechnol. 36(7): 566–569.ArticlePubMedPMC
- Bauer E, Zimmermann J, Baldini F, Thiele I, Kaleta C. 2017. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput Biol. 13(5): e1005544.ArticlePubMedPMC
- Beghini F, McIver LJ, Blanco-Miguez A, Dubois L, Asnicar F, et al. 2021. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 10: e65088.ArticlePubMedPMC
- Beura S, Kundu P, Das AK, Ghosh A. 2024. Genome-scale community modelling elucidates the metabolic interaction in Indian type-2 diabetic gut microbiota. Sci Rep. 14(1): 17259.ArticlePubMedPMCPDF
- Birkel GW, Ghosh A, Kumar VS, Weaver D, Ando D, et al. 2017. The JBEI quantitative metabolic modeling library (jQMM): a python library for modeling microbial metabolism. BMC Bioinform. 18(1): 205.ArticlePDF
- Brunk E, Sahoo S, Zielinski DC, Altunkaya A, Drager A, et al. 2018. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat Biotechnol. 36(3): 272–281.ArticlePubMedPMCPDF
- Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, et al. 2020. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 48(D1): D445–D453.ArticlePubMedPDF
- Chan SHJ, Simons MN, Maranas CD. 2017. SteadyCom: Predicting microbial abundances while ensuring community stability. PLoS Comput Biol. 13(5): e1005539.ArticlePubMedPMC
- Chen CZ, Li P, Liu L, Sun YJ, Ju WM, et al. 2024. Seasonal variations of microbial communities and viral diversity in fishery-enhanced marine ranching sediments: insights into metabolic potentials and ecological interactions. Microbiome. 12(1): 209.ArticlePubMedPMCPDF
- Christians U, Klawitter J, Hornberger A, Klawitter J. 2011. How unbiased is non-targeted metabolomics and is targeted pathway screening the solution? Curr Pharm Biotechnol. 12(7): 1053–1066.ArticlePubMed
- Cottret L, Frainay C, Chazalviel M, Cabanettes F, Gloaguen Y, et al. 2018. MetExplore: collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 46(W1): W495–W502.ArticlePubMedPMC
- Cupples AM, Li Z, Wilson FP, Ramalingam V, Kelly A. 2022. In silico analysis of soil, sediment and groundwater microbial communities to predict biodegradation potential. J Microbiol Methods. 202: 106595.ArticlePubMed
- Dopson M, Rezaei Somee M, Gonzalez-Rosales C, Lui LM, Turner S, et al. 2024. Novel candidate taxa contribute to key metabolic processes in Fennoscandian Shield deep groundwaters. ISME Commun. 4(1): ycae113.ArticlePubMedPMCPDF
- Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, et al. 2020. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 38(6): 685–688.ArticlePubMedPMCPDF
- Dukovski I, Bajic D, Chacon JM, Quintin M, Vila JCC, et al. 2021. A metabolic modeling platform for the computation of microbial ecosystems in time and space (COMETS). Nat Protoc. 16(11): 5030–5082.ArticlePubMedPMCPDF
- Fernandes P, Sharma Y, Zulqarnain F, McGrew B, Shrivastava A, et al. 2023. Identifying metabolic shifts in Crohn's disease using 'omics-driven contextualized computational metabolic network models. Sci Rep. 13(1): 203.ArticlePubMedPMCPDF
- Gonzalez JM, Aranda B. 2023. Microbial growth under limiting conditions-future perspectives. Microorganisms. 11(7): 1641.ArticlePubMedPMC
- Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, et al. 2013. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Res. 41(D1): D387–395.ArticlePubMed
- Heinken A, Hertel J, Acharya G, Ravcheev DA, Nyga M, et al. 2023. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat Biotechnol. 41(9): 1320–1331.ArticlePubMedPMCPDF
- Heinken A, Thiele I. 2022. Microbiome Modelling Toolbox 2.0: efficient, tractable modelling of microbiome communities. Bioinformatics. 38(8): 2367–2368.ArticlePubMedPMCPDF
- Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, et al. 2019. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat Protoc. 14(3): 639–702.ArticlePubMedPMCPDF
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, et al. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 28(9): 977–982.ArticlePDF
- Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. 2023. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1): D587–D592.ArticlePubMedPDF
- Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, et al. 2019. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 20(4): 1085–1093.ArticlePubMedPDF
- King ZA, Lu J, Drager A, Miller P, Federowicz S, et al. 2016. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 44(D1): D515–522.ArticlePubMed
- Kumar M, Ji B, Babaei P, Das P, Lappa D, et al. 2018. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab Eng. 49: 128–142.ArticlePubMedPMC
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, et al. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 31(9): 814–821.ArticlePubMedPMCPDF
- Le V, Quinn TP, Tran T, Venkatesh S. 2020. Deep in the bowel: Highly interpretable neural encoder-decoder networks predict gut metabolites from gut microbiome. BMC Genomics. 21(Suppl 4): 256.ArticlePubMedPMCPDF
- Li M, Hu X, Ni T, Ni Y, Xue D, et al. 2024. Comparative genomic analyses of the genus Robertmurraya and proposal of the novel species Robertmurraya mangrovi sp. nov., isolated from mangrove soil. Antonie van Leeuwenhoek. 118(1): 22.ArticlePubMedPDF
- Louca S, Doebeli M. 2015. Calibration and analysis of genome-based models for microbial ecology. Elife. 4: e08208.ArticlePubMedPMCPDF
- Machado D, Andrejev S, Tramontano M, Patil KR. 2018. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46(15): 7542–7553.ArticlePubMedPMC
- Mallick H, Franzosa EA, McLver LJ, Banerjee S, Sirota-Madi A, et al. 2019. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat Commun. 10(1): 3136.ArticlePubMedPMCPDF
- Marshall DD, Powers R. 2017. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog Nucl Magn Reson Spectrosc. 100: 1–16.ArticlePubMedPMC
- Mendes-Soares H, Mundy M, Soares LM, Chia N. 2016. MMinte: an application for predicting metabolic interactions among the microbial species in a community. BMC Bioinformatics. 17(1): 343.ArticlePubMedPMCPDF
- Muller E, Shiryan I, Borenstein E. 2024. Multi-omic integration of microbiome data for identifying disease-associated modules. Nat Commun. 15(1): 2621.ArticlePubMedPMCPDF
- Nhu VH, Mohammadi A, Shahabi H, Shirzadi A, Al-Ansari N, et al. 2020. Monitoring and assessment of water level fluctuations of the Lake Urmia and its environmental consequences using multitemporal Landsat 7 ETM+ images. Int J Environ Res Public Health. 17(12): 4210.ArticlePubMedPMC
- Noecker C, Eng A, Muller E, Borenstein E. 2022. MIMOSA2: a metabolic network-based tool for inferring mechanism-supported relationships in microbiome-metabolome data. Bioinformatics. 38(6): 1615–1623.ArticlePubMedPMCPDF
- Noronha A, Modamio J, Jarosz Y, Guerard E, Sompairac N, et al. 2019. The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47(D1): D614–D624.ArticlePubMedPDF
- Northen TR, Kleiner M, Torres M, Kovacs AT, Nicolaisen MH, et al. 2024. Community standards and future opportunities for synthetic communities in plant-microbiota research. Nat Microbiol. 9(11): 2774–2784.ArticlePubMedPDF
- Ostos I, Florez-Pardo LM, Camargo C. 2024. A metagenomic approach to demystify the anaerobic digestion black box and achieve higher biogas yield: a review. Front Microbiol. 15: 1437098.ArticlePubMedPMC
- Parras-Molto M, Aguirre de Carcer D. 2020. A comprehensive human minimal gut metagenome extends the host's metabolic potential. Microb Genom. 6(11): mgen000466.Article
- Proffitt C, Bidkhori G, Lee S, Tebani A, Mardinoglu A, et al. 2022. Genome-scale metabolic modelling of the human gut microbiome reveals changes in the glyoxylate and dicarboxylate metabolism in metabolic disorders. iScience. 25(7): 104513.ArticlePubMedPMC
- Raes EJ, Karsh K, Sow SLS, Ostrowski M, Brown MV, et al. 2021. Metabolic pathways inferred from a bacterial marker gene illuminate ecological changes across South Pacific frontal boundaries. Nat Commun. 12(1): 2213.ArticlePubMedPMCPDF
- Reiman D, Layden BT, Dai Y. 2021. MiMeNet: Exploring microbiome-metabolome relationships using neural networks. PLoS Comput Biol. 17(5): e1009021.ArticlePubMedPMC
- Seaver SMD, Liu F, Zhang Q, Jeffryes J, Faria JP, et al. 2021. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 49(D1): D575–D588.ArticlePubMedPDF
- Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, et al. 2015. Quantifying diet-Induced metabolic changes of the human gut microbiome. Cell Metab. 22(2): 320–331.ArticlePubMed
- Shoaie S, Nielsen J. 2014. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front Genet. 5: 86.ArticlePubMedPMC
- Shtossel O, Koren O, Shai I, Rinott E, Louzoun Y. 2024. Gut microbiome-metabolome interactions predict host condition. Microbiome. 12(1): 24.ArticlePubMedPMCPDF
- Swainston N, Smallbone K, Hefzi H, Dobson PD, Brewer J, et al. 2016. Recon 2.2: from reconstruction to model of human metabolism. Metabolomics. 12: 109.ArticlePubMedPMCPDF
- Thiele I, Palsson BO. 2010. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 5(1): 93–121.ArticlePubMedPMCPDF
- Vuckovic D. 2012. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal Bioanal Chem. 403(6): 1523–1548.ArticlePubMedPDF
- Wang H, Marcisauskas S, Sanchez BJ, Domenzain I, Hermansson D, et al. 2018. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput Biol. 14(10): e1006541.ArticlePubMedPMC
- Yin X, Altman T, Rutherford E, West KA, Wu Y, et al. 2020. A comparative evaluation of tools to predict metabolite profiles from microbiome sequencing data. Front Microbiol. 11: 595910.ArticlePubMedPMC
- Zhang B, Magnaye KM, Stryker E, Moltzau-Anderson J, Porsche CE, et al. 2024. Sustained mucosal colonization and fecal metabolic dysfunction by Bacteroides associates with fecal microbial transplant failure in ulcerative colitis patients. Sci Rep. 14(1): 18558.ArticlePubMedPMCPDF
- Zheng J, Sun Q, Zhang M, Liu C, Su Q, et al. 2024. Noninvasive, microbiome-based diagnosis of inflammatory bowel disease. Nat Med. 30: 3555–3567.ArticlePubMedPMCPDF
- Zhou Z, Tran PQ, Breister AM, Liu Y, Kieft K, et al. 2022. METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome. 10(1): 33.ArticlePubMedPMCPDF
- Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, et al. 2011. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 5(2): 305–316.ArticlePubMedPDF
- Zomorrodi AR, Maranas CD. 2012. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 8(2): e1002363.ArticlePubMedPMC
- Zorrilla F, Buric F, Patil KR, Zelezniak A. 2021. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Res. 49(21): e126.ArticlePubMedPMCPDF
References
Supplementary Information
References
Citations
- Next‐Generation Eco‐Omics: Integrating Microbial Function Into Predictive Ecosystem Models
Kulmani Mehar, Kamakshi Priya K, Amit Prakash Sen, Ravi Kumar Paliwal, Bhavan Kumar M., Aravindan Munusamy Kalidhas, Tapas Kumar Mohapatra, Aseel Samrat, Ravikumar Jayabal
Biotechnology and Applied Biochemistry.2026; 73(3): 1667. CrossRef - The Role of Genitourinary Microbiome in Male Cancer Etiology and Progression: Insights from Next-Generation Sequencing and Meta-Omics
Pooja Tiwary, Krishil Oswal, Ryan Varghese
Société Internationale d’Urologie Journal.2026; 7(1): 9. CrossRef - Bioinformatics in Antifungal Design: Strategies To Overcome Resistance from a Proteomic Perspective
Diego Romário-Silva, Edja Maria Melo de Brito Costa, Joanilda Paolla Raimundo Silva, Letícia Targino Campos, Vitória Marina Abrantes Batista, Camila Vital de Araújo, Sonaly Lima Albino, Arthur Gabriel Corrêa de Farias, Igor José dos Santos Nascimento, Ric
Current Fungal Infection Reports.2026;[Epub] CrossRef - 16S-Pipeline: A comprehensive web-based platform for end-to-end 16S rRNA amplicon sequencing analysis
Tatsuya Unno
Journal of Microbiology.2026; 64(5): e2603014. CrossRef - Advances in Enzymatic Production of Prebiotic Oligosaccharides from Agro-Industrial Waste: A Critical Review and Industrial Framework
Slim Smaoui
Foods.2026; 15(12): 2156. CrossRef - Activity-guided discovery of antibiotic-transforming bacteria from environmental microbiomes using D-amino acid–assisted fluorescence-activated cell sorting
Yi Liu, Kai-Li Wang, Yu-Qi Hong, Ye Yuan, Hua Wang, Yi-Qun Chen, Sheng-Song Yu, Zi-Xuan Lu, Yuan Pan, Ting-Ting Zhu
Environmental Pollution.2026; 405: 128591. CrossRef - An inferential ceiling in nanomaterial-assisted phytoremediation studies: Insights from a semi-systematic review of functional evidence in soil microbial communities
Ottavia Pinto, Marco Contin, Luca Marchiol
Applied Soil Ecology.2026; 225: 107199. CrossRef - Microbiome–metabolite signaling networks in gastrointestinal disease: systems biology, network rewiring, and precision therapeutics
Yahya A. Almutawif, Hamza M. A. Eid
Archives of Microbiology.2026;[Epub] CrossRef - Naringenin: From sustainable biosynthesis to gut microbiota-mediated bioactivation and systemic health outcomes
Shutong Liu, Tian Gong, Chenxu Zhao, Chaoqun Zhang, Yanhui Han, Hang Xiao, Yonghong Meng
Trends in Food Science & Technology.2026; 176: 105914. CrossRef - Microbiota, chronic inflammation, and health: The promise of inflammatome and inflammatomics for precision medicine and health care
Huan Zhang, Bing Jun Yang Lee, Tong Wang, Xuesong Xiang, Yafang Tan, Yanping Han, Yujing Bi, Fachao Zhi, Xin Wang, Fang He, Seppo J. Salminen, Baoli Zhu, Ruifu Yang
hLife.2025; 3(7): 307. CrossRef - Study on the Rhizosphere Soil Microbial Diversity of Five Common Orchidaceae Species in the Transitional Zone Between Warm Temperate and Subtropical Regions
Jingjing Du, Shengqian Guo, Xiaohang Li, Zhonghu Geng, Zhiliang Yuan, Xiqiang Song
Diversity.2025; 17(9): 605. CrossRef - Bioengineered Skin Microbiome: The Next Frontier in Personalized Cosmetics
Cherelle Atallah, Ayline El Abiad, Marita El Abiad, Mantoura Nakad, Jean Claude Assaf
Cosmetics.2025; 12(5): 205. CrossRef - Computational Metagenomics: State of the Art
Marco Antonio Pita-Galeana, Martin Ruhle, Lucía López-Vázquez, Guillermo de Anda-Jáuregui, Enrique Hernández-Lemus
International Journal of Molecular Sciences.2025; 26(18): 9206. CrossRef - Rotation of Corydalis yanhusuo with different crops enhances its quality and soil nutrients: a multi-dimensional analysis of rhizosphere microecology
Jia Liu, Qiang Yuan, Kejie Zhang, Xiaoxiao Sheng, Zixuan Zhu, Ning Sui, Hui Wang
BMC Plant Biology.2025;[Epub] CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article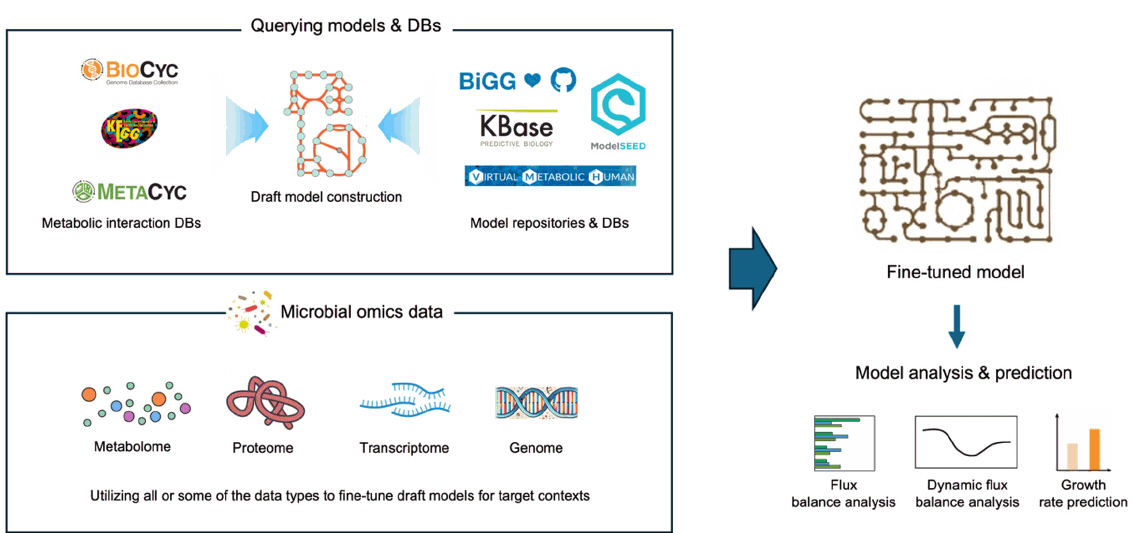
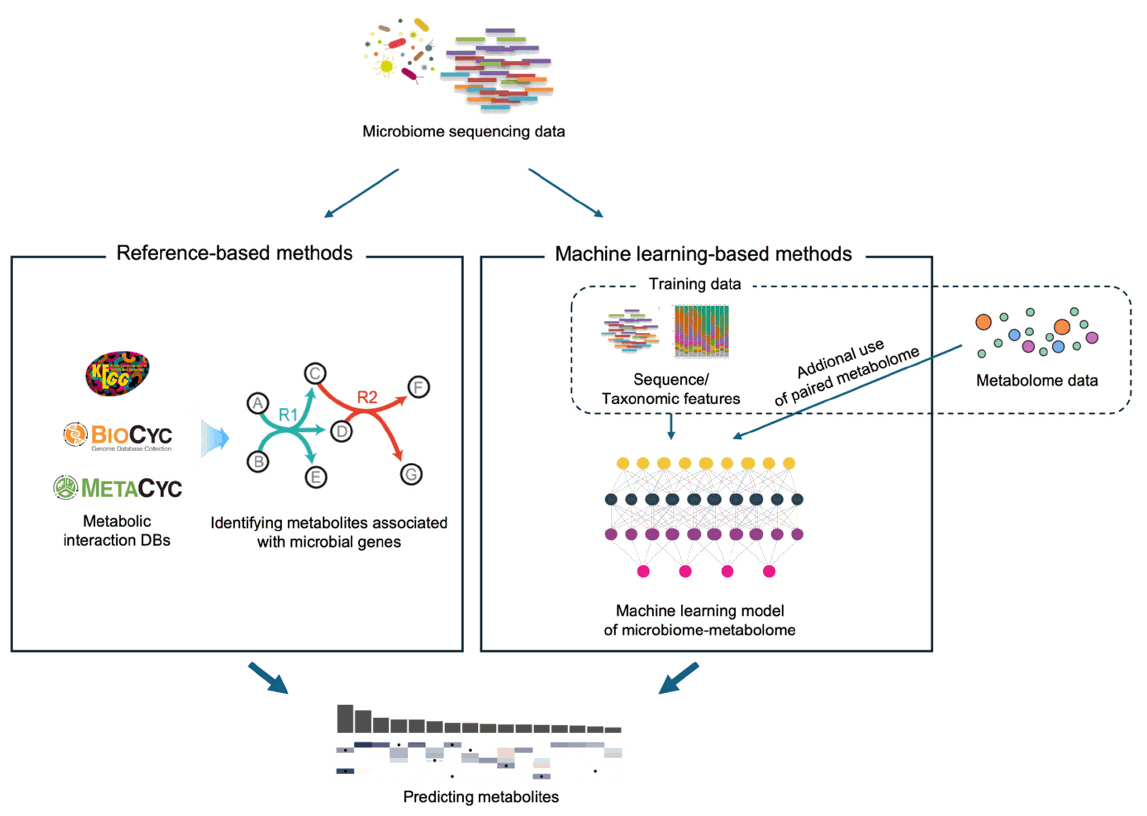


Fig. 1.
Fig. 2.
| Name | Purpose/Functionality | Key features |
|---|---|---|
| AGORA2 ( |
Personalized and predictive modeling | Models of 7,302 microbial strains |
| Model repository | Information on 98 drugs and relevant enzymes | |
| BacArena ( |
Individual-based metabolic modeling of microbial communities | Integrates FBA with individual-based modeling |
| Modeling spatial and temporal dynamics | ||
| BiGG ( |
Repository for GEMs | 77 manually curated GEMs |
| Knowledge integration | Supporting various model formats | |
| Community collaboration | Supporting web API | |
| CarveMe ( |
Fast reconstruction of GEMs for microbial species and communities | Top-down approach using a universal model for scalable model generation |
| Automated gap-filling for improved growth phenotype predictions | ||
| COBRA ( |
Constraint-based modeling of biochemical networks | Extensive support for FBA and omics data integration |
| High-performance solvers for multi-scale and genome-scale models | ||
| COMETS ( |
Dynamic simulation of microbial community interactions | Spatially structured dFBA |
| Supports Python and MATLAB interfaces for customized simulations | ||
| DyMMM ( |
Simulating interactions and competition in microbial communities under dynamic conditions | Integrates genome-scale models for multi-species interactions |
| Predicts community dynamics under varying environmental conditions | ||
| jQMM ( |
Modeling microbial metabolism and analyzing omics data | Combines FBA and 13C metabolic flux analysis |
| Uses 13C labeling data for genome-scale model constraints | ||
| KBase ( |
Data sharing, integration, and analysis for systems biology | Diverse data integration (genomes, biochemistry) |
| Web-based interface with data provenance | ||
| MCM ( |
Modeling multi-species microbial communities with genome-based metabolic models | Statistical parameter calibration with experimental data |
| dFBA for metabolic interaction simulation | ||
| metaGEM ( |
Reconstruction of GEMs from metagenome | End-to-end pipeline for community-level metabolic interaction simulations |
| Generates personalized metabolic models from metagenome-assembled genomes (MAGs) | ||
| MetExplore ( |
Collaborative curation and exploration of metabolic networks | Data mapping for multi-omics integration |
| Sub-network extraction and interactive visualization | ||
| Microbiome Modeling Toolbox ( |
Efficient modeling and analysis of microbiome communities | Parallelized generation of personalized microbiome models |
| Visualization and statistical analysis for model comparison | ||
| MMinte ( |
Predicts metabolic interactions among microbial species in a community | Pairwise interaction analysis under different metabolic conditions |
| Modular interface with independent functionalities for flexibility | ||
| ModelSEED ( |
High-throughput generation and optimization of GEMs | Automated reconstruction pipeline from genome annotation to draft models |
| Integrates gap-filling for biomass production and growth simulation | ||
| OptCom ( |
Multi-level optimization for modeling metabolic interactions in microbial communities | Balances individual VS. community fitness criteria |
| Captures various interaction types (positive, negative) for multiple species | ||
| RAVEN ( |
Reconstruction and analysis of GEMs | Supports de novo model reconstruction using KEGG and MetaCyc databases |
| Integration with COBRA Toolbox for compatibility and bi-directional model conversion | ||
| SteadyCom ( |
Predicting microbial community composition and maintaining steady-state growth | Ensures constant community growth rate across all species |
| Supports flux variability analysis to explore metabolic flexibility | ||
| VMH ( |
Integration of models with extrinsic factors such as nutrition and disease | Extensive data coverage (Recon3D human model, 818 microbial models, disease/nutrition information) |
| Method type | Name | Data requirements | Advantages | Limitations |
|---|---|---|---|---|
| ML-based | LOCATE ( |
Paired microbiome (16S or metagenomics) and metabolomics data | Latent representation and low data requirement for training | Limited cross-dataset generalization |
| Reference-based | Mangosteen ( |
Microbiome sequencing data | Utilizes curated databases | Limited by database coverage |
| ML-based | MelonnPan ( |
Amplicon or metagenomic sequencing data, paired with metabolomic data for training | Predicts metabolomic profiles from metagenomic data | Requires training data and limited generalization |
| ML-based | MiMeNet ( |
Paired microbiome (metagenomic taxonomic/functional) and metabolome data | Improves prediction via multivariate learning | Performance depends on dataset size |
| Reference-based | MIMOSA2 ( |
Paired microbiome (16S or metagenomics) and metabolomics data | Infers mechanistic microbe-metabolite links | Limited to environments represented in reference databases |
| Name | Approach | Input data | Unique features |
|---|---|---|---|
| bioBakery ( |
Reference-based, assembly-independent profiling | Metagenomic and metatranscriptomic sequences | Integrates taxonomic, strain-level, functional, and phylogenetic profiling |
| METABOLIC ( |
High-throughput metabolic and biogeochemical profiling | Genomes from isolates, metagenome-assembled genomes, or single-cell genomes | Community-scale functional networks |
| MintTea ( |
Identification of multi-omic modules | Taxonomic, Functional, Metabolome profiles | Integration of multi-modal data and identifying predictive modules |
| PICRUSt2 ( |
Phylogenetic placement and hidden state prediction | 16S rRNA gene sequences | ASV compatibility, supports custom databases |
Table 1.
Table 2.
Table 3.
TOP

