- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(2); 2025 > Article
-
Full article
Small molecule kinase inhibitor altiratinib inhibits brain cyst forming bradyzoites of Toxoplasma gondii - Yeong Hoon Kim1, Hye-Jin Ahn2, Hwa Sun Kim3, Ho-Woo Nam2,*
-
Journal of Microbiology 2025;63(2):e2409001.
DOI: https://doi.org/10.71150/jm.2409001
Published online: February 27, 2025
1Department of Ophthalmology, College of Medicine, The Catholic University of Korea, Seoul 06591, Republic of Korea
2Department of Parasitology, College of Medicine, The Catholic University of Korea, Seoul 06591, Republic of Korea
3Department of Family Medicine, Veterans Health Service Medical Center, Seoul 05368, Republic of Korea
- *Correspondence Ho-Woo Nam howoo@catholic.ac.kr
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 3,398 Views
- 82 Download
ABSTRACT
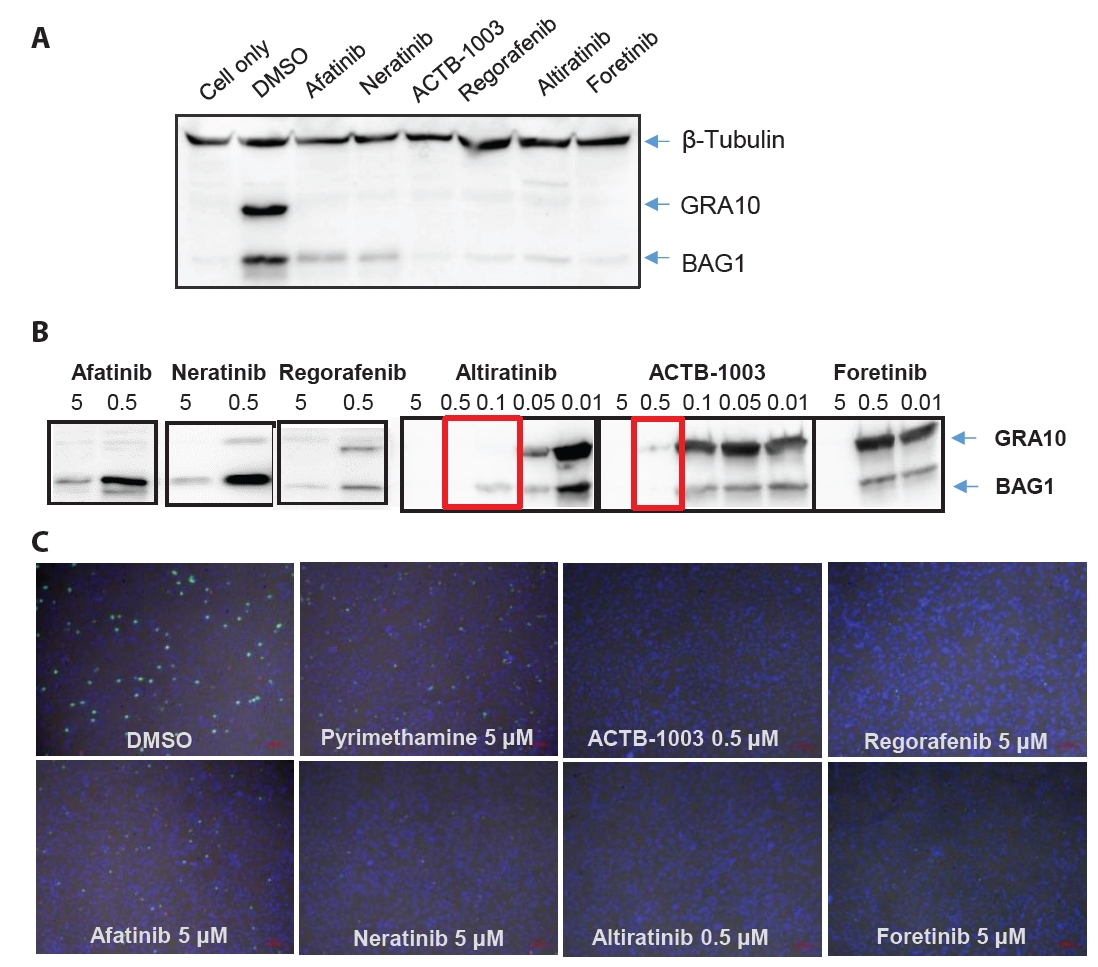
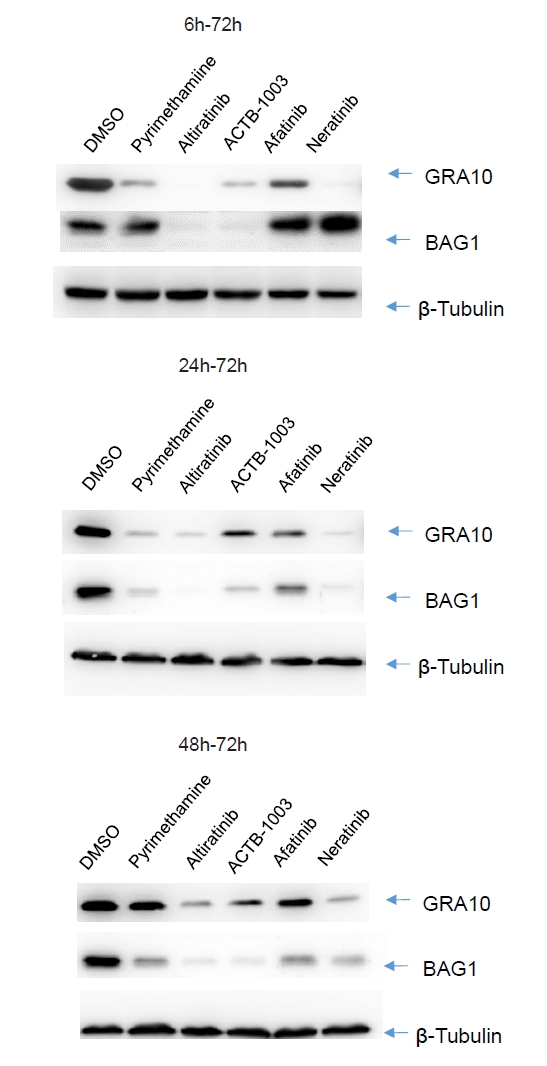
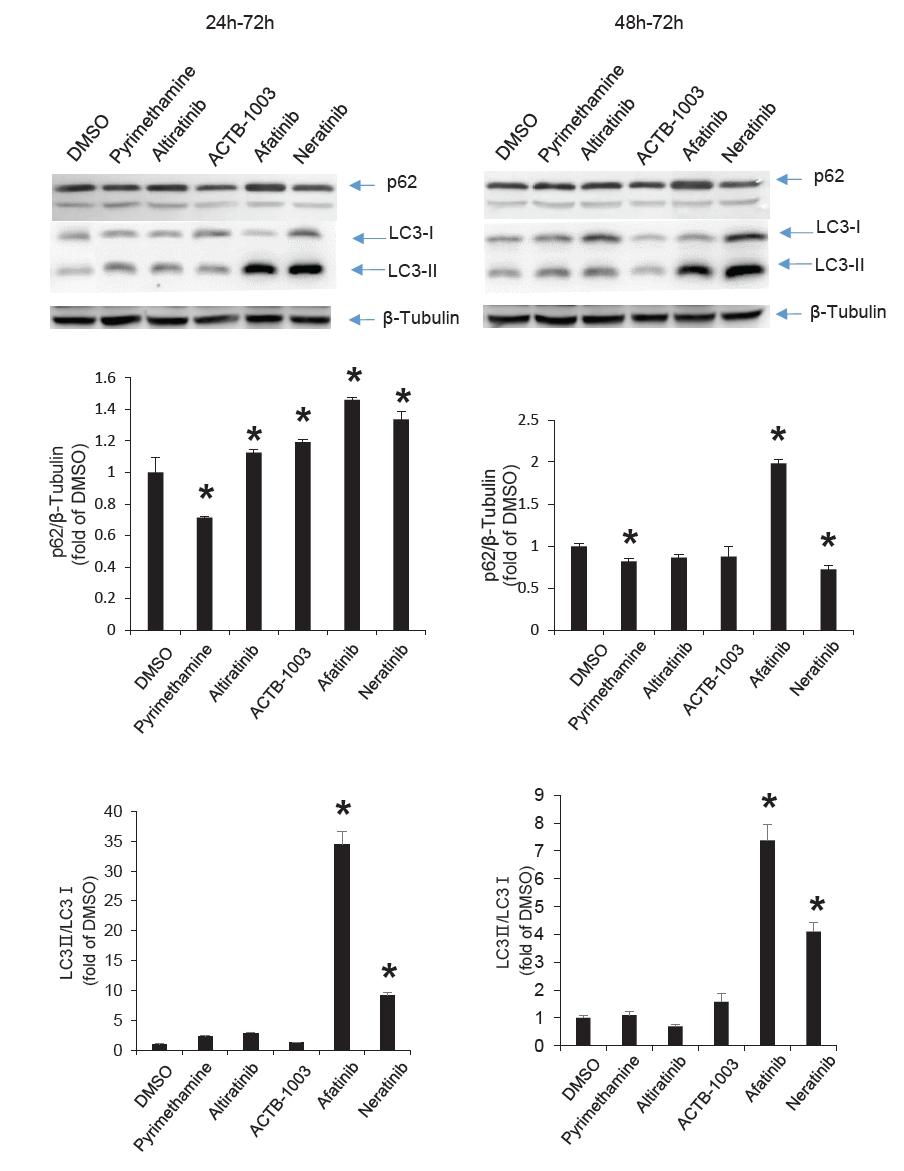
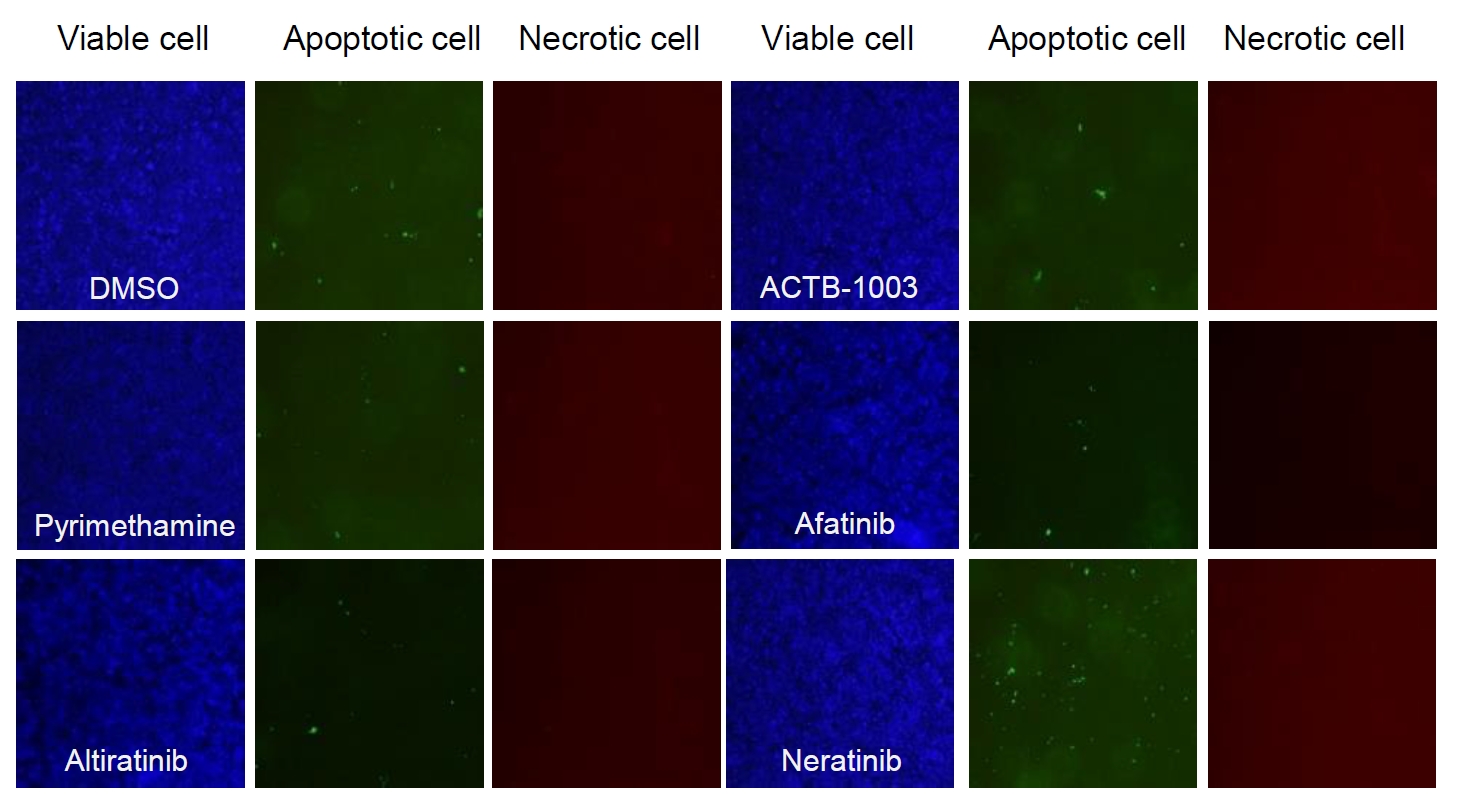
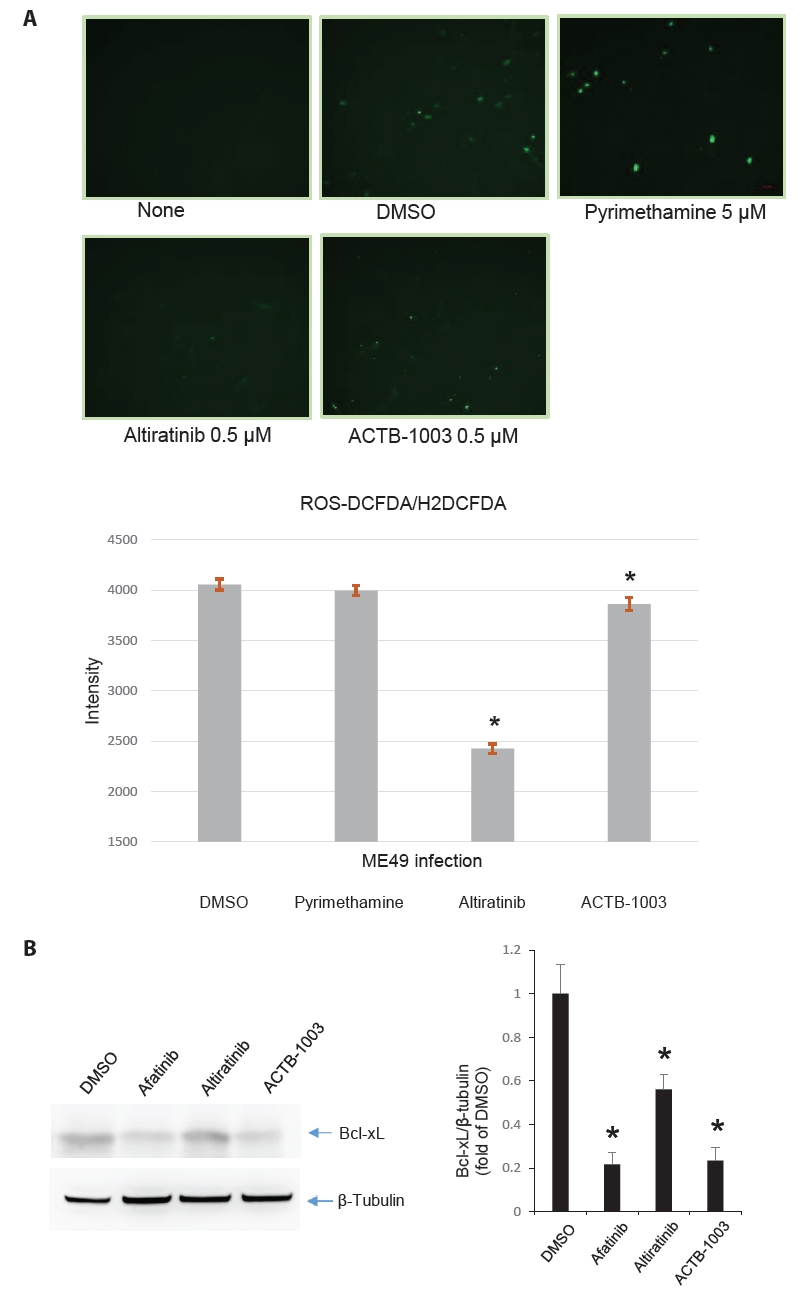
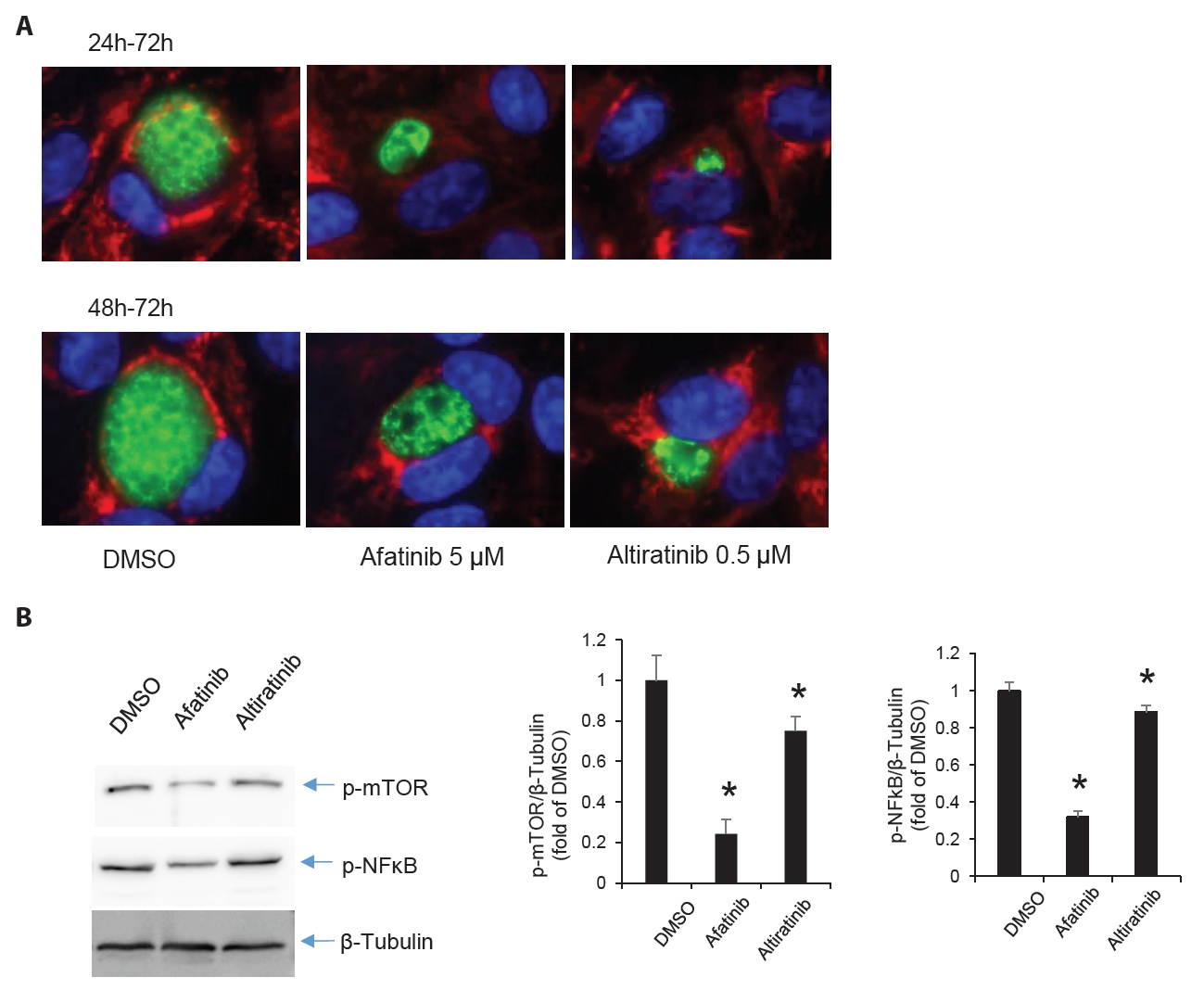
- Chronic toxoplasmosis is caused by Toxoplasma gondii bradyzoites. This study assessed six candidate small molecule kinase inhibitors (SMKIs) against bradyzoites (ME49 strain), the reactivated form of the parasite resulting from the rupture of brain cysts. Bradyzoites were obtained from mouse brain cysts, cultured in ARPE-19 cells, and treated with afatinib and neratinib (HER2/HER4 inhibitors), ACTB-1003 and regorafenib (VEGFR-2 inhibitors), or altiratinib and foretinib (c-MET inhibitors). The effects on the growth of T. gondii were analyzed by western blot and immunofluorescence assay. Changes in the host cells were assessed using markers for cell viability, apoptosis, necrosis, and autophagy. All inhibitors blocked the growth of bradyzoites, although afatinib was less effective. Afatinib enhanced autophagy signals, while ACTB-1003 and neratinib affected mitochondrial biosynthesis and mitophagy. Altiratinib demonstrated an effect against bradyzoites at the lowest concentration with minimal impact on the host cells. It may be effective in blocking the reactivation of brain cysts in immunodeficiency patients caused by bradyzoites.
Introduction
Materials and Methods
Results
Discussion
Acknowledgments
The work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (no. NRF-2022R1F1A1063799).
Conflict of Interest
The authors have no financial nor any other competing conflicts of interest to declare.
Ethical Statements
All procedures and handling of mice were conducted under an approved protocol by the Institutional Animal Care and Use Committee (IACUC) at the School of Medicine, The Catholic University of Korea (CUMC-2023–0012–01), which adhered to the regulations set under the Korean National Animal Protection Act.
Supplementary Information
Fig. S1.
Fig. S2.
- Araujo FG, Huskinson-Mark J, Gutteridge WE, Remington JS. 1992. In vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against the cyst form of Toxoplasma gondii. Antimicrob Agents Chemother. 36(2): 326–330. ArticlePubMedPMCLink
- Carey KL, Westwood NJ, Mitchison TJ, Ward GE. 2004. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc Natl Acad Sci USA. 101(20): 7433–7438. ArticlePubMedPMC
- Carrolo M, Giordano S, Cabrita-Santos L, Corso S, Vigario AM, et al. 2003. Hepatocyte growth factor and its receptor are required for malaria infection. Nat Med. 9(11): 1363–1369. ArticlePubMedPDF
- Cerutti A, Blanchard N, Besteiro S. 2020. The bradyzoite: A key developmental stage for the persistence and pathogenesis of toxoplasmosis. Pathogens. 9(3): 234–254. ArticlePubMedPMC
- Child MA, Hall CI, Beck JR, Ofori LO, Albrow VE, et al. 2013. Small-molecule inhibition of a depalmitoylase enhances Toxoplasma host-cell invasion. Nat Chem Biol. 9(9): 651–656. ArticlePubMedPMCPDF
- Choi HG, Gao FF, Zhou W, Sun PR, Yuk JM, et al. 2020. The role of PI3K/AKT pathway and NADPH oxidase 4 in host ROS manipulation by Toxoplasma gondii. Korean J Parasitol. 58(3): 237–247. ArticlePubMedPMC
- Danilkovitch-Miagkova A, Zbar B. 2002. Dysregulation of MET receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 109(7): 863–867. ArticlePubMedPMC
- DeLong MJ. 1998. Apoptosis: A modulator of cellular homeostasis and disease states. Ann N Y Acad Sci. 842: 82–90. ArticlePubMed
- Dubey JP, Lindsay DS, Speer CA. 1998. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev. 11(2): 267–299. ArticlePubMedPMCLink
- Ferguson DJ, Hutchison WM. 1987. The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch A Pathol Anat Histopathol. 411(1): 39–43. ArticlePubMedPDF
- Frenkel J. 1956. Pathogenesis of toxoplasmosis and of infections with organisms resembling Toxoplasma. Ann N Y Acad Sci. 64(2): 215–251. Article
- Gormley PD, Pavesio CE, Minnasian D, Lightman S. 1998. Effects of drug therapy on Toxoplasma cysts in an animal model of acute and chronic disease. Invest Ophthalmol Vis Sci. 39(7): 1171–1175. PubMed
- Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, et al. 1999. VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res. 247(2): 495–504. ArticlePubMed
- Hashizume H, Falcon BL, Kuroda T, Baluk P, Coxon A, et al. 2010. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 70(6): 2213–2223. ArticlePubMedPMCPDF
- Hua QQ, Lin XJ, Xiang SP, Jiang LY, Cai JH, et al. 2023. Two small-molecule inhibitors of Toxoplasma gondii proliferation in vitro. Front Cell Infect Microbiol. 13: 1145824.ArticlePubMedPMC
- Hubbard SR, Till JH. 2000. Protein tyrosine kinase structure and function. Annu Rev Biochem. 69: 373–398. ArticlePubMed
- Jacobs L, Remington JS, Melton ML. 1960. The resistance of the encysted form of Toxoplasma gondii. J Parasitol. 46: 11–21. ArticlePubMed
- Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, et al. 2013. Gene expression profile identifies tyrosine kinase MET as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 19(7): 1773–1783. ArticlePubMedPMC
- Jakubczyk K, Dec K, Kaldunska J, Kawczuga D, Kochman J, et al. 2020. Reactive oxygen species: Sources, functions, oxidative damage. Pol Merkur Lekarski. 48(284): 124–127. ArticlePubMed
- Kaur H, Sarmah D, Veeresh P, Datta A, Kalia K, et al. 2021. Endovascular stem cell therapy post stroke rescues neurons from endoplasmic reticulum stress-induced apoptosis by modulating brain-derived neurotrophic factor/Tropomyosin receptor kinase B signaling. ACS Chem Neurosci. 12(19): 3745–3759. ArticlePubMed
- Kim I, Moon SO, Han CY, Pak YK, Moon SK, et al. 2001. The angiopoietin-Tie2 system in coronary artery endothelium prevents oxidized low-density lipoprotein-induced apoptosis. Cardiovasc Res. 49(4): 872–881. ArticlePubMed
- Kim YH, Bhatt L, Ahn HJ, Yang Z, Lee WK, et al. 2017. Suppressors for human epidermal growth factor receptor 2/4 (HER2/4): A new family of anti-toxoplasmic agents in ARPE-19 cells. Korean J Parasitol. 55(5): 491–503. ArticlePubMedPMC
- Kortagere S. 2012. Screening for small molecule inhibitors of Toxoplasma gondii. Expert Opin Drug Discov. 7(12): 1193–1206. ArticlePubMed
- Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. 2015. Effective inhibition of MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 34(2): 144–153. ArticlePubMedPDF
- Leiriao P, Albuquerque SS, Corso S, van Gemert GJ, Sauerwein RW, et al. 2005. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell Microbiol. 7(4): 603–609. ArticlePubMed
- Lepri S, Nannetti G, Muratore G, Cruciani G, Ruzziconi R, et al. 2014. Optimization of small-molecule inhibitors of influenza virus polymerase: From thiophene-3-carboxamide to polyamido scaffolds. J Med Chem. 57(10): 4337–4350. ArticlePubMed
- Levine B, Klionsky DJ. 2004. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 6(4): 463–477. ArticlePubMed
- Levitzki A, Gazit A. 1995. Tyrosine kinase inhibition: An approach to drug development. Science. 267(5205): 1782–1788. ArticlePubMed
- Martinez-Gil L, Alamares-Sapuay JG, Ramana Reddy MV, Goff PH, Premkumar Reddy E, et al. 2013. A small molecule multi-kinase inhibitor reduces influenza A virus replication by restricting viral RNA synthesis. Antiviral Res. 100(1): 29–37. ArticlePubMed
- McCannel CA, Holland GN, Helm CJ, Cornell PJ, Winston JV, et al. 1996. Causes of uveitis in the general practice of ophthalmology. UCLA community-based uveitis study group. Am J Ophthalmol. 121(1): 35–46. ArticlePubMed
- Miman O, Kusbeci OY, Aktepe OC, Cetinkaya Z. 2010. The probable relation between Toxoplasma gondii and Parkinson's disease. Neurosci Lett. 475(2): 129–131. ArticlePubMed
- Molan A, Nosaka K, Hunter M, Wang W. 2019. Global status of Toxoplasma gondii infection: Systematic review and prevalence snapshots. Trop Biomed. 36(4): 898–925. PubMed
- Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet. 363(9425): 1965–1976. ArticlePubMed
- Muniz-Feliciano L, Van Grol J, Portillo JA, Liew L, Liu B, et al. 2013. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog. 9(5): e1003809. ArticlePubMedPMC
- Ngo HM, Zhou Y, Lorenzi H, Wang K, Kim TK, et al. 2017. Toxoplasma modulates signature pathways of human epilepsy, neurodegeneration, and cancer. Sci Rep. 7(1): 11496.ArticlePubMedPMC
- Passelli K, Prat-Luri B, Merlot M, Goris M, Mazzone M, et al. 2022. The c-MET receptor tyrosine kinase contributes to neutrophil-driven pathology in cutaneous leishmaniasis. PLoS Pathog. 18(2): e1010247. ArticlePubMedPMC
- Peixoto L, Chen F, Harb OS, Davis PH, Beiting DP, et al. 2010. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe. 8(3): 208–218. ArticlePubMedPMC
- Piao Y, Park SY, Henry V, Smith BD, Tiao N, et al. 2016. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro Oncol. 18(9): 1230–1241. ArticlePubMedPMC
- Pradhan E, Bhandari S, Gilbert RE, Stanford M. 2016. Antibiotics versus no treatment for Toxoplasma retinochoroiditis. Cochrane Database Syst Rev. 2016(5): CD002218.ArticlePubMedPMC
- Puthiyakunnon S, He X, Boddu S, Huang SH, Cao H. 2017. C-MET inhibitors are potential novel therapeutic agents against Listeria monocytogenes infection through blocking the bacteria entry into nonphagocytic cells. Curr Top Med Chem. 17(3): 278–289. ArticlePubMed
- Rutaganira FU, Barks J, Dhason MS, Wang Q, Lopez MS, et al. 2017. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and recurrence of central nervous system disease by Toxoplasma gondii. J Med Chem. 60(24): 9976–9989. ArticlePubMedPMC
- Smith BD, Kaufman MD, Leary CB, Turner BA, Wise SC, et al. 2015. Altiratinib inhibits tumor growth, invasion, angiogenesis, and microenvironment-mediated drug resistance via balanced inhibition of MET, TIE2, and VEGFR2. Mol Cancer Ther. 14(8): 2023–2034. ArticlePubMedPDF
- Soete M, Camus D, Dubremetz JF. 1994. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp Parasitol. 78(4): 361–370. ArticlePubMed
- Sun K, Bröms J, Lavander M, Gurram BK, Enquist PA, et al. 2014. Screening for inhibition of Vibrio cholerae VipA-VipB interaction identifies small-molecule compounds active against type VI secretion. Antimicrob Agents Chemother. 58(7): 4123–4130. ArticlePubMedPMCLink
- Sun PR, Gao FF, Choi HG, Zhou W, Yuk JM, et al. 2019. Dipenyleneiodonium induces growth inhibition of Toxoplasma gondii through ROS induction in ARPE-19 cells. Korean J Parasitol. 57(2): 83–92. ArticlePubMedPMC
- Swale C, Bellini V, Bowler MW, Flore N, Brenier-Pinchart MP, et al. 2022. Altiratinib blocks Toxoplasma gondii and Plasmodium falciparum development by selectively targeting a spliceosome kinase. Sci Transl Med. 14(659): eabn3231.ArticlePubMed
- Tedford E, McConkey G. 2017. Neurophysiological changes induced by chronic Toxoplasma gondii infection. Pathogens. 6(2): 19.ArticlePubMedPMC
- Tuck AB, Park M, Sterns EE, Boag A, Elliott BE. 1996. Coexpression of hepatocyte growth factor and receptor (MET) in human breast carcinoma. Am J Pathol. 148(1): 225–232. PubMedPMC
- Woods JR Jr, Plessinger MA, Fantel A. 1998. An introduction to reactive oxygen species and their possible roles in substance abuse. Obstet Gynecol Clin North Am. 25(2): 219–236. ArticlePubMed
- Yang Z, Ahn HJ, Park YH, Nam HW. 2016. Afatinib reduces STAT6 signaling of host ARPE-19 cells infected with Toxoplasma gondii. Korean J Parasitol. 54(1): 31–38. ArticlePubMedPMC
- Yu CP, Chen BC, Chou YC, Hsieh CJ, Lin FH. 2023. The epidemiology of patients with toxoplasmosis and its associated risk factors in Taiwan during the 2007-2020 period. PLoS One. 18(8): e0290769. ArticlePubMedPMC
- Zhang J, Yang PL, Gray NS. 2009. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 9(1): 28–39. ArticlePubMedPDF
References
Supplementary Information
References
Citations
 ePub Link
ePub Link Cite this Article
Cite this Article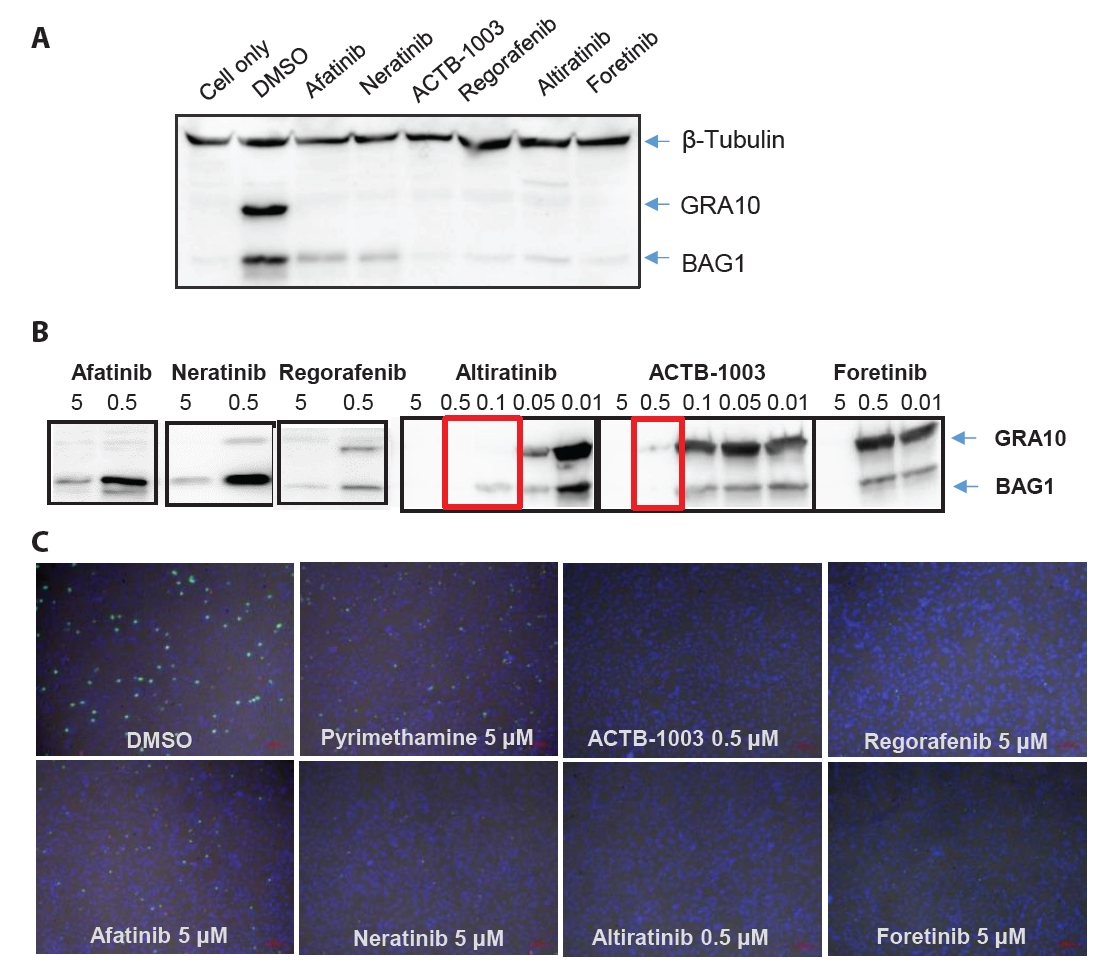
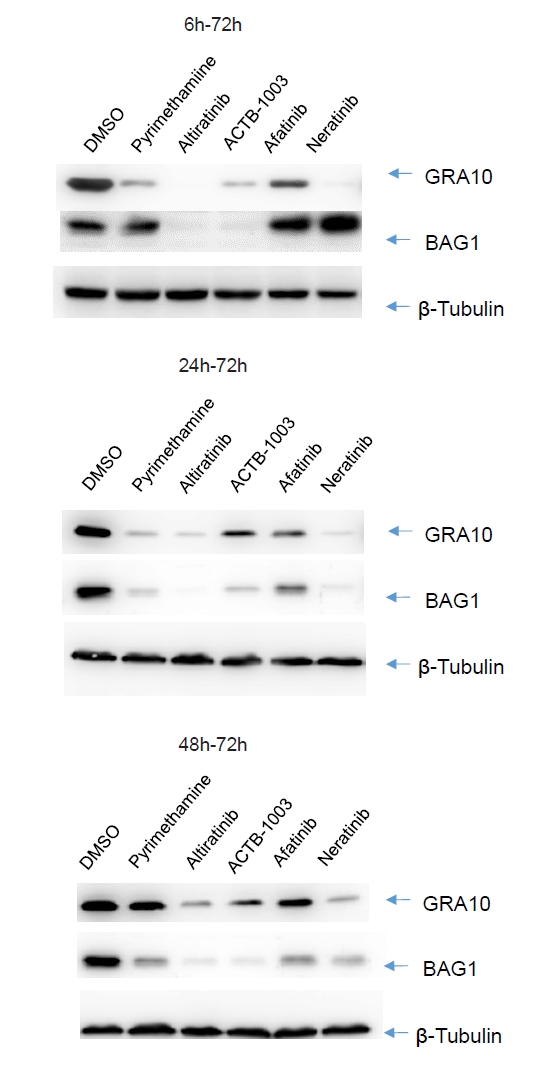
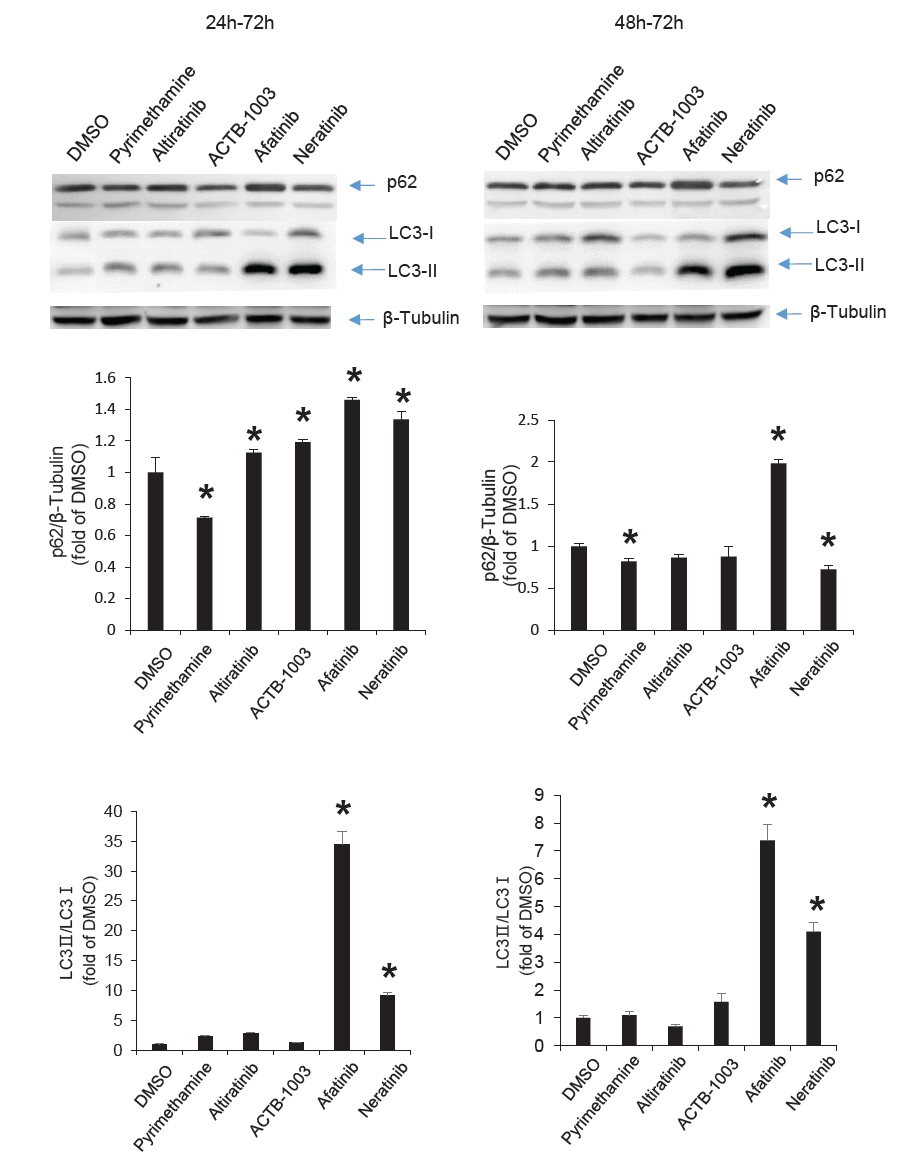
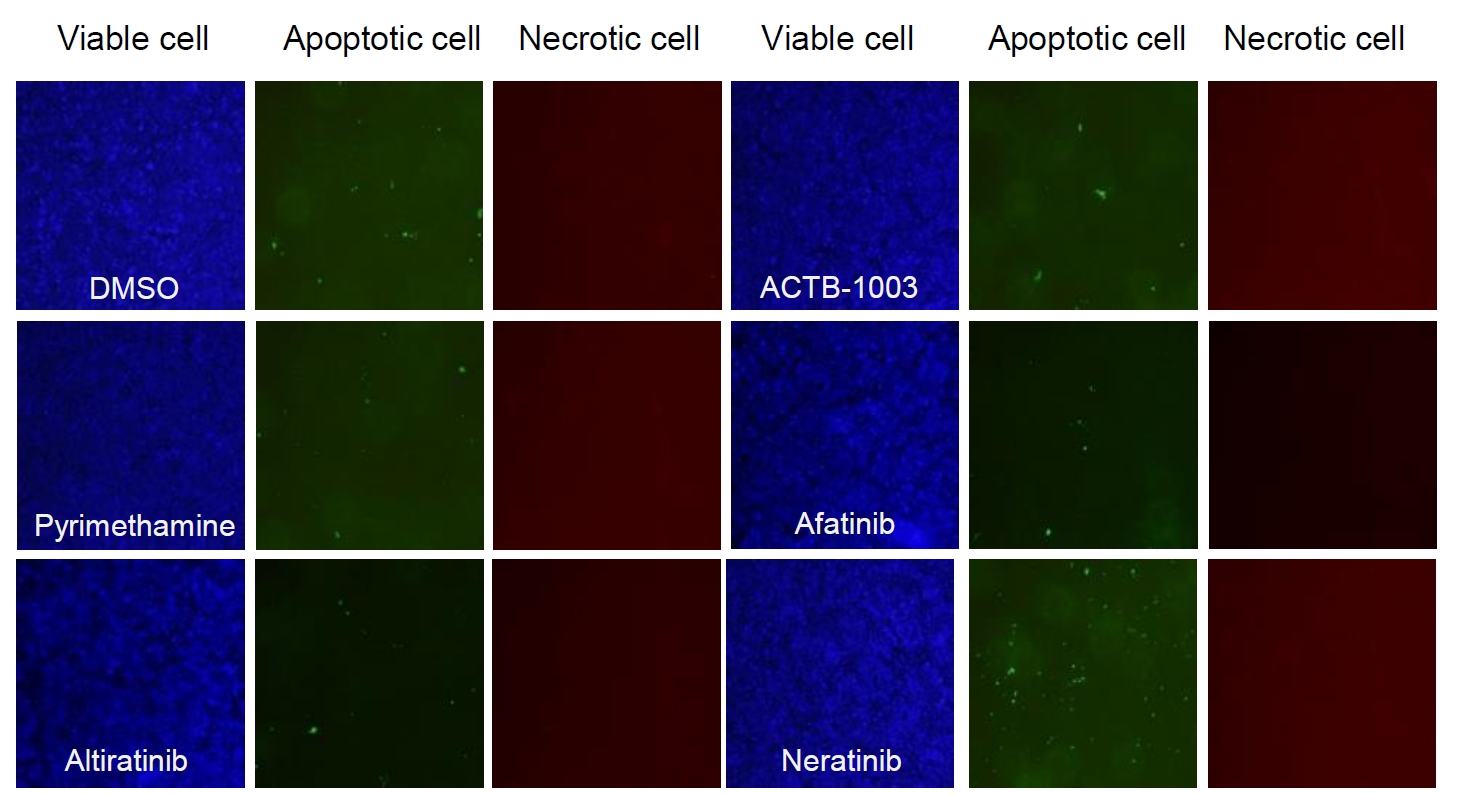
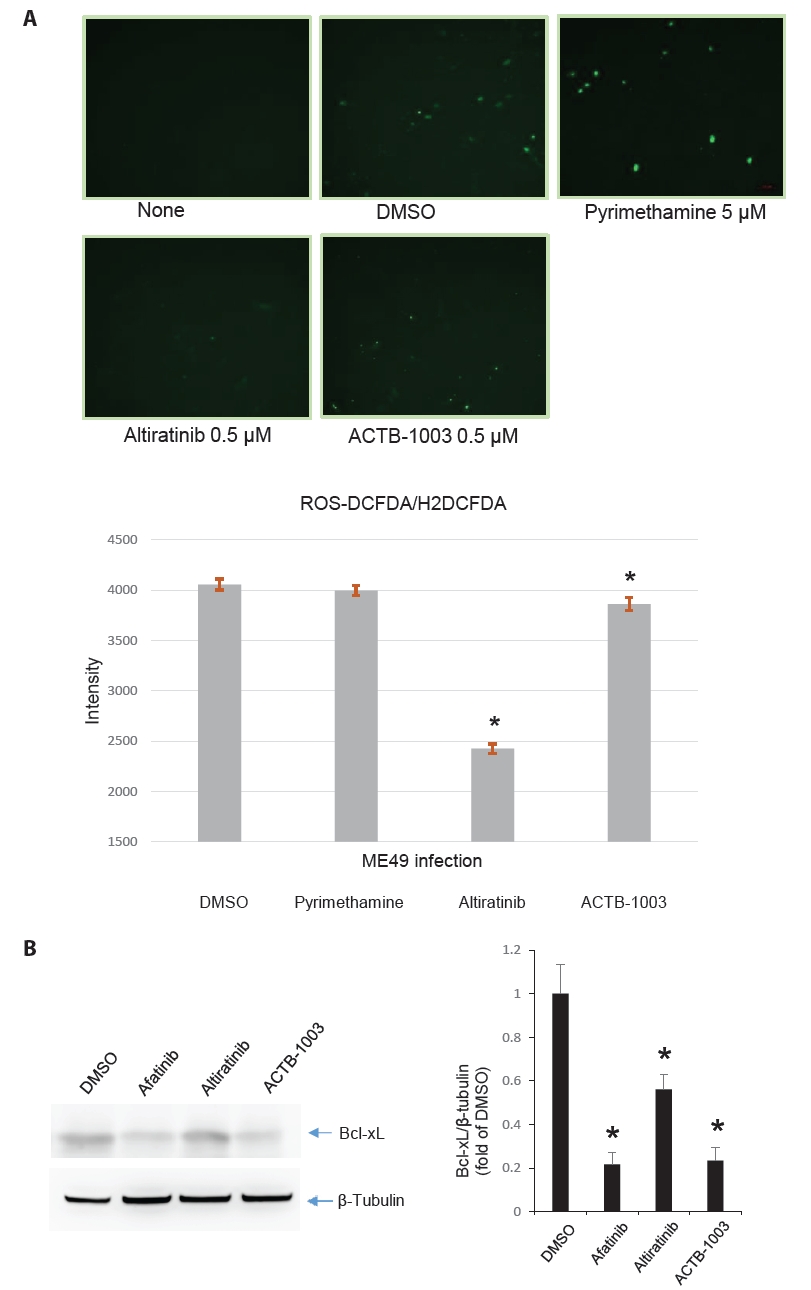
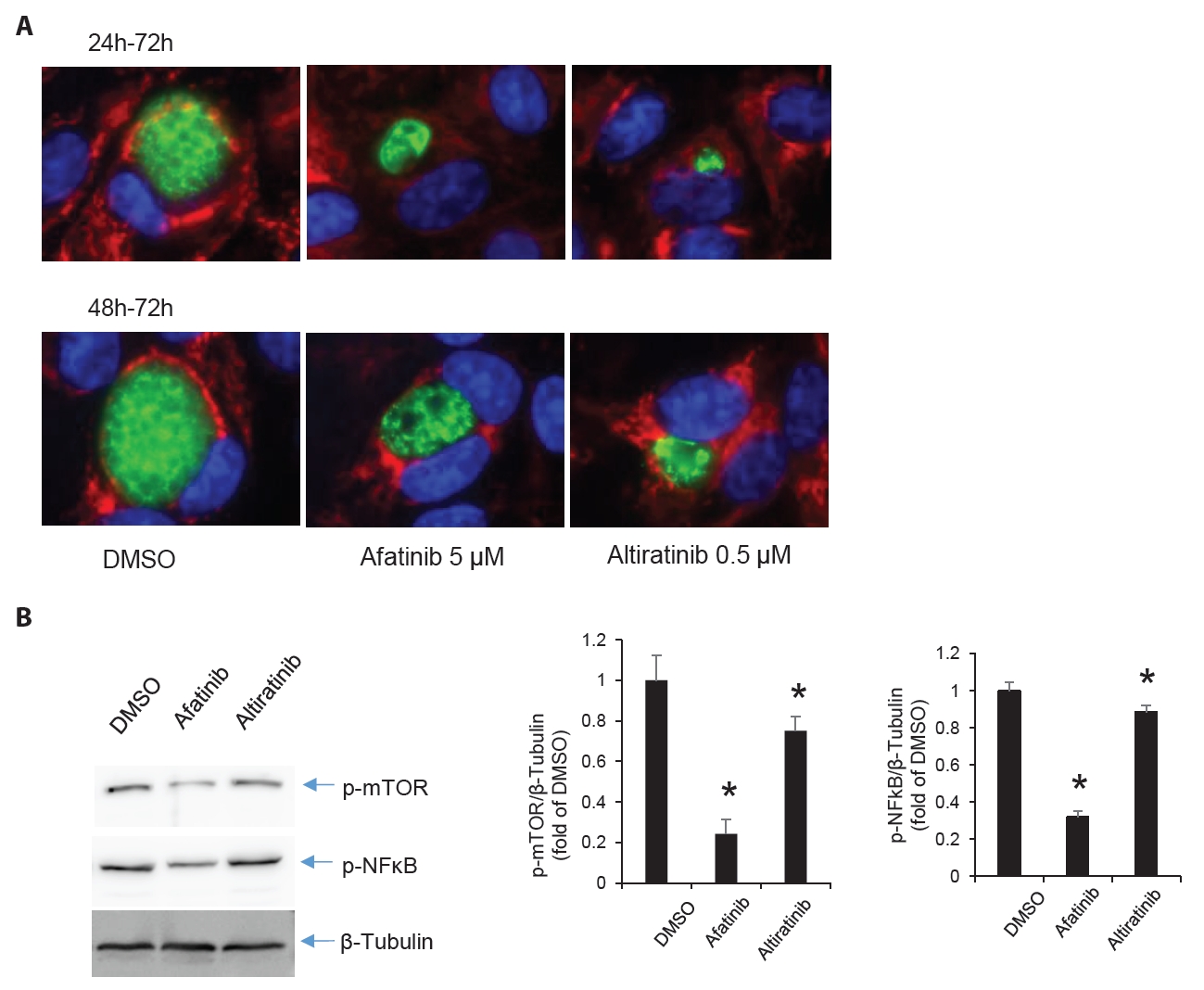






Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
TOP

