- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(9); 2025 > Article
-
Full article
Efficient CRISPR-based genome editing for inducible degron systems to enable temporal control of protein function in large double-stranded DNA virus genomes - Kihye Shin1,2, Eui Tae Kim1,2,*
-
Journal of Microbiology 2025;63(9):e2504008.
DOI: https://doi.org/10.71150/jm.2504008
Published online: August 29, 2025
1Department of Microbiology and Immunology, Jeju National University College of Medicine, Jeju 63241, Republic of Korea
2Jeju Research Center for Natural Medicine, Jeju National University Core Research Institute, Jeju 63241, Republic of Korea
- *Correspondence Eui Tae Kim tae@jejunu.ac.kr
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- CRISPR-Cas9-based gene editing enables precise genetic modifications. However, its application to human cytomegalovirus (HCMV) remains challenging due to the large size of the viral genome and the essential roles of key regulatory genes. Here, we establish an optimized CRISPR-Cas9 system for precise labeling and functional analysis of HCMV immediate early (IE) genes. By integrating a multifunctional cassette encoding an auxin-inducible degron (AID), a self-cleaving peptide (P2A), and GFP into the viral genome via homology-directed repair (HDR), we achieved efficient knock-ins without reliance on bacterial artificial chromosome (BAC) cloning, a labor-intensive and time-consuming approach. We optimized delivery strategies, donor template designs, and component ratios to enhance HDR efficiency, significantly improving knock-in success rates. This system enables real-time fluorescent tracking and inducible protein degradation, allowing temporal control of essential viral proteins through auxin-mediated depletion. Our approach provides a powerful tool for dissecting the dynamic roles of viral proteins throughout the HCMV life cycle, facilitating a deeper understanding of viral pathogenesis and potential therapeutic targets.
Introduction
Materials and Methods
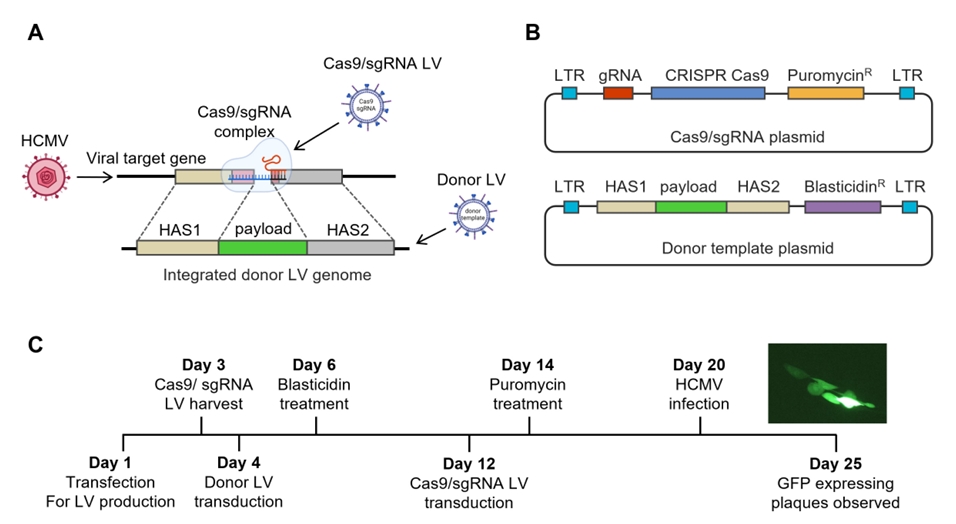
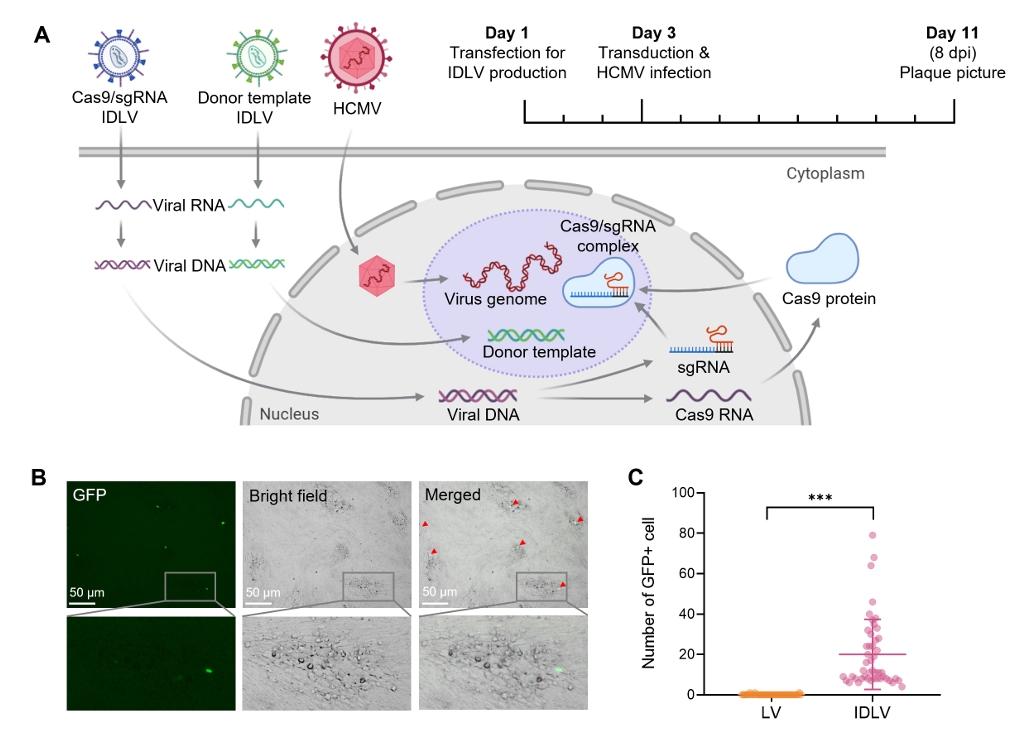
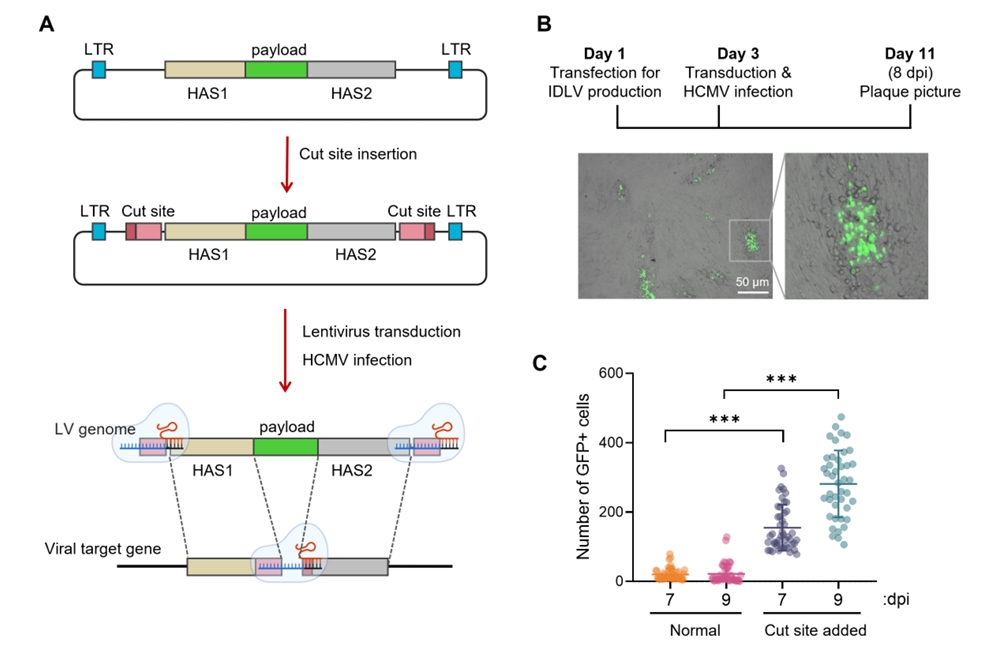
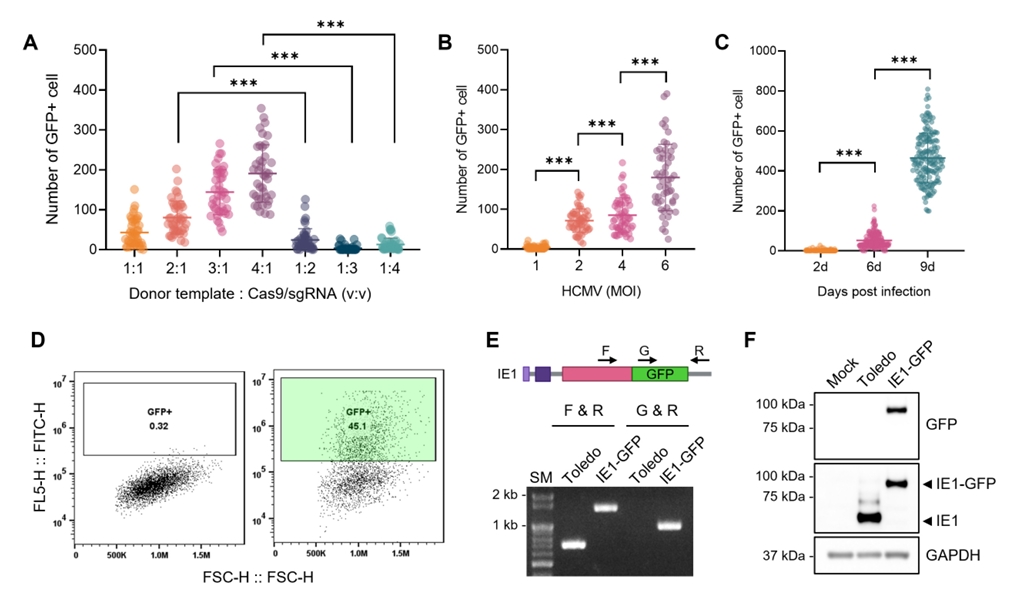
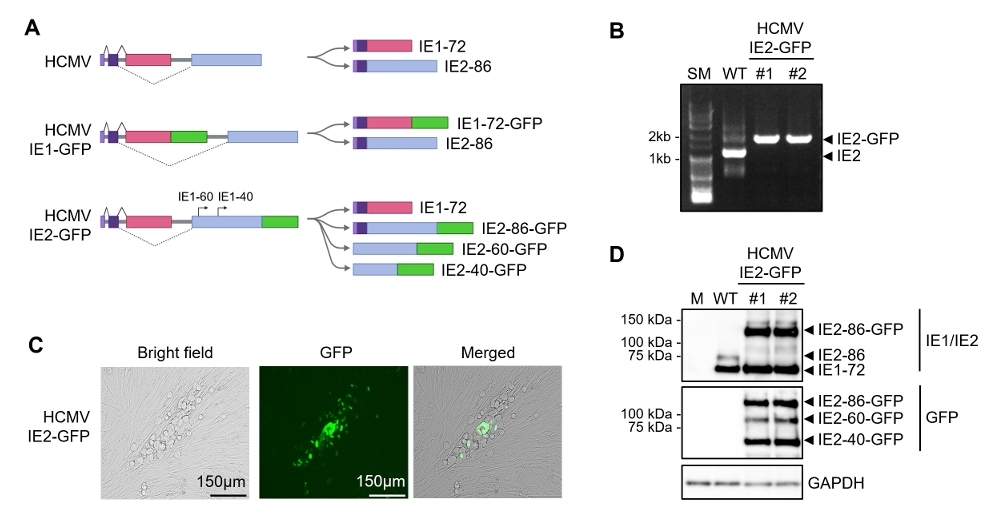
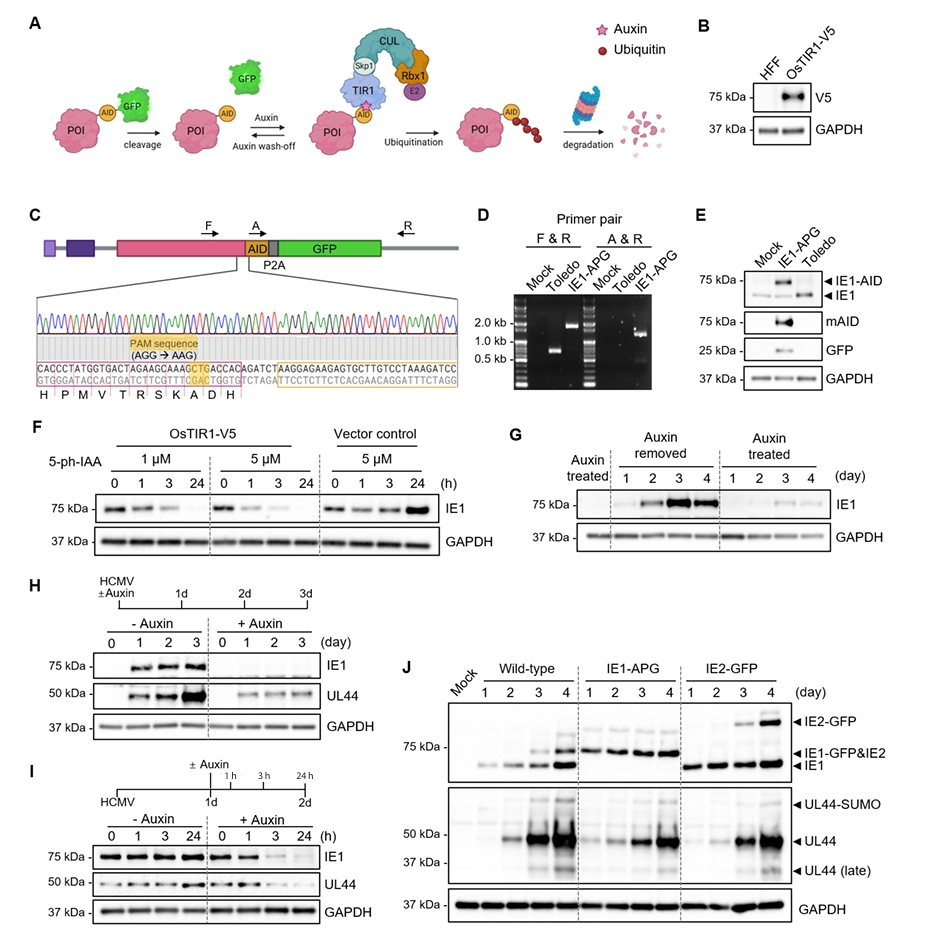
Results
Discussion
Acknowledgments
We thank members of the Kim lab for insightful discussion and input. We are also grateful to Professor Jin-Hyun Ahn (Sungkyunkwan University School of Medicine) for generously providing the HCMV Toledo strain and rabbit-derived antiserum against IE1.
Conflict of Interest
The authors declare no potential conflicts of interest.
Funding
This work was supported by the National Research Foundation of Korea (NRF), with grants from the Ministry of Science and ICT (RS-2024-00352590), Ministry of Education (RS-2023-00270936), and the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (RS-2022-KH129726, RS-2024-00438990).
- Adamson CS, Nevels MM. 2020. Bright and early: inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses. 12: 110.ArticlePubMedPMC
- Ahn JH, Brignole EJ 3rd, Hayward GS. 1998. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol Cell Biol. 18: 4899–4913. ArticlePubMedPMCLink
- Collins-McMillen D, Rak M, Buehler JC, Igarashi-Hayes S, Kamil JP, et al. 2019. Alternative promoters drive human cytomegalovirus reactivation from latency. Proc Natl Acad Sci USA. 116: 17492–17497. ArticlePubMedPMC
- Forte E, Zhang Z, Thorp EB, Hummel M. 2020. Cytomegalovirus latency and reactivation: an intricate interplay with the host immune response. Front Cell Infect Microbiol. 10: 186.ArticlePubMedPMC
- Gaur M, Leavitt AD. 1998. Mutations in the human immunodeficiency virus type 1 integrase D,D(35)E motif do not eliminate provirus formation. J Virol. 72: 4678–4685. ArticlePubMedPMCLink
- Griffiths P, Reeves M. 2021. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat Rev Microbiol. 19: 759–773. ArticlePubMedPMCPDF
- Kim YE, Ahn JH. 2015. Positive role of promyelocytic leukemia protein in type I interferon response and its regulation by human cytomegalovirus. PLoS Pathog. 11: e1004785. ArticlePubMedPMC
- Leal AF, Herreno-Pachón AM, Benincore-Flórez E, Karunathilaka A, Tomatsu S. 2024. Current strategies for increasing knock-in efficiency in CRISPR/Cas9-based approaches. Int J Mol Sci. 25: 2456.ArticlePubMedPMC
- Liao H, Wu J, VanDusen NJ, Li Y, Zheng Y. 2024. CRISPR-Cas9-mediated homology-directed repair for precise gene editing. Mol Ther Nucleic Acids. 35: 102344.ArticlePubMedPMC
- Lin G, Blissard GW. 2002. Analysis of an Autographa californica nucleopolyhedrovirus lef-11 knockout: LEF-11 is essential for viral DNA replication. J Virol. 76: 2770–2779. ArticlePubMedPMCLink
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, et al. 2017. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep. 7: 2193.ArticlePubMedPMCPDF
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. 2009. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 6: 917–922. ArticlePubMedPDF
- Rana R, Biegalke BJ. 2014. Human cytomegalovirus UL34 early and late proteins are essential for viral replication. Viruses. 6: 476–488. ArticlePubMedPMC
- Reeves M, Sinclair J. 2013. Regulation of human cytomegalovirus transcription in latency: beyond the major immediate-early promoter. Viruses. 5: 1395–1413. ArticlePubMedPMC
- Shin HJ, Kim YE, Kim ET, Ahn JH. 2012. The chromatin-tethering domain of human cytomegalovirus immediate-early (IE) 1 mediates associations of IE1, PML and STAT2 with mitotic chromosomes, but is not essential for viral replication. J Gen Virol. 93: 716–721. ArticlePubMed
- Van Damme E, Van Loock M. 2014. Functional annotation of human cytomegalovirus gene products: an update. Front Microbiol. 5: 218.ArticlePubMedPMC
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, et al. 2017. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell. 168: 890–903.e15. ArticlePubMedPMC
- Warden C, Tang Q, Zhu H. 2011. Herpesvirus BACs: past, present, and future. J Biomed Biotechnol. 2011: 124595.ArticlePubMedPMCPDF
- Yesbolatova A, Saito Y, Kitamoto N, Makino-Itou H, Ajima R, et al. 2020. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat Commun. 11: 5701.ArticlePubMedPMCPDF
References
Supplementary Information
References
Citations
- Viral genome editing methods and applications in the CRISPR era
Kihye Shin, Eui Tae Kim, Herman W. Favoreel
Journal of Virology.2026;[Epub] CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article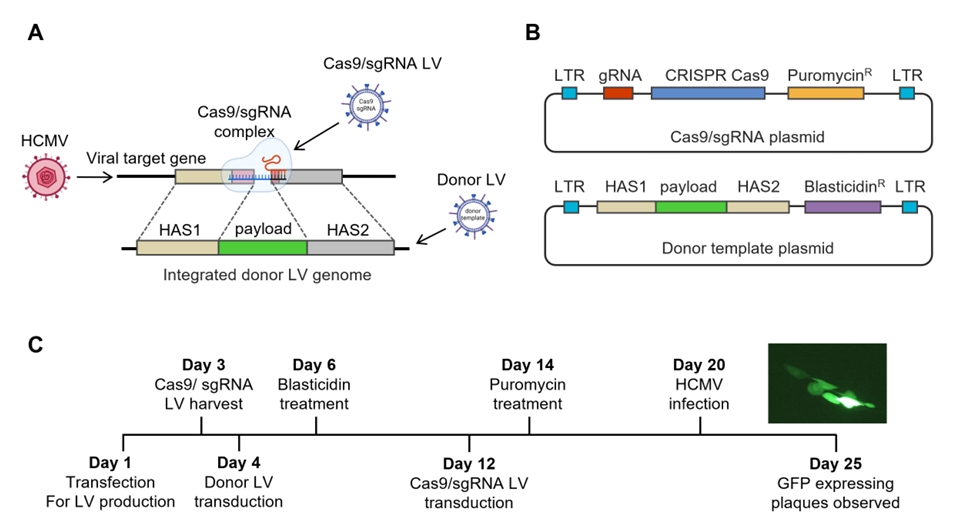
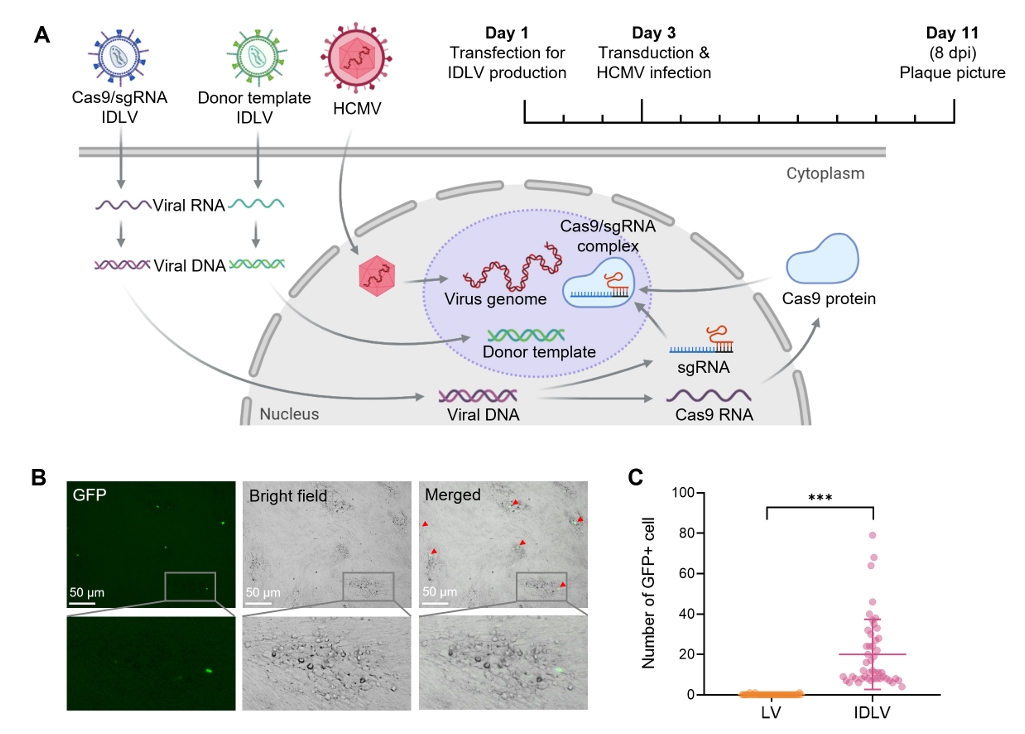
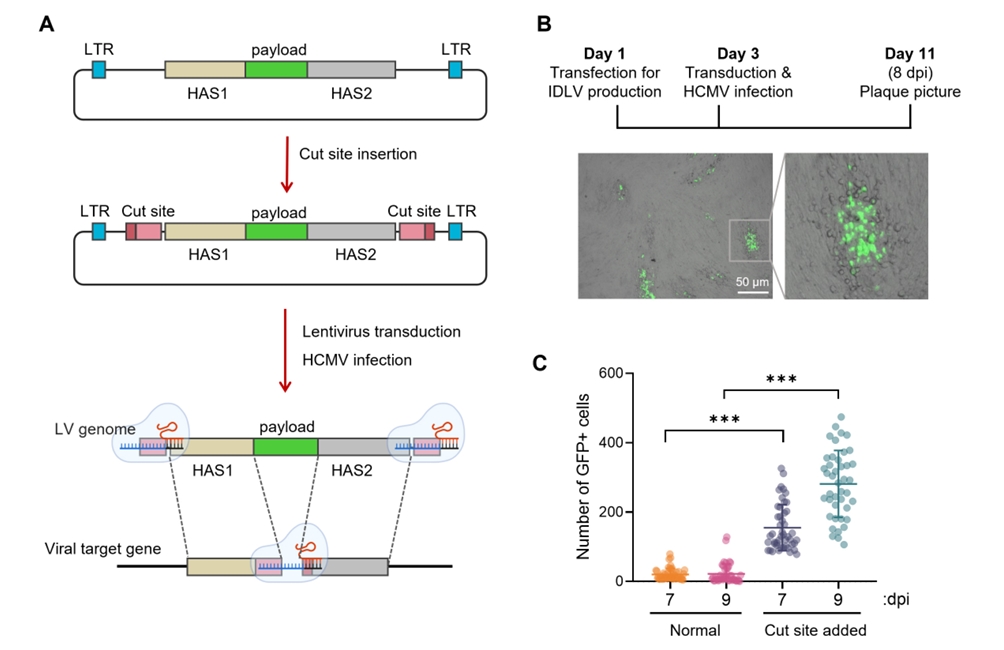
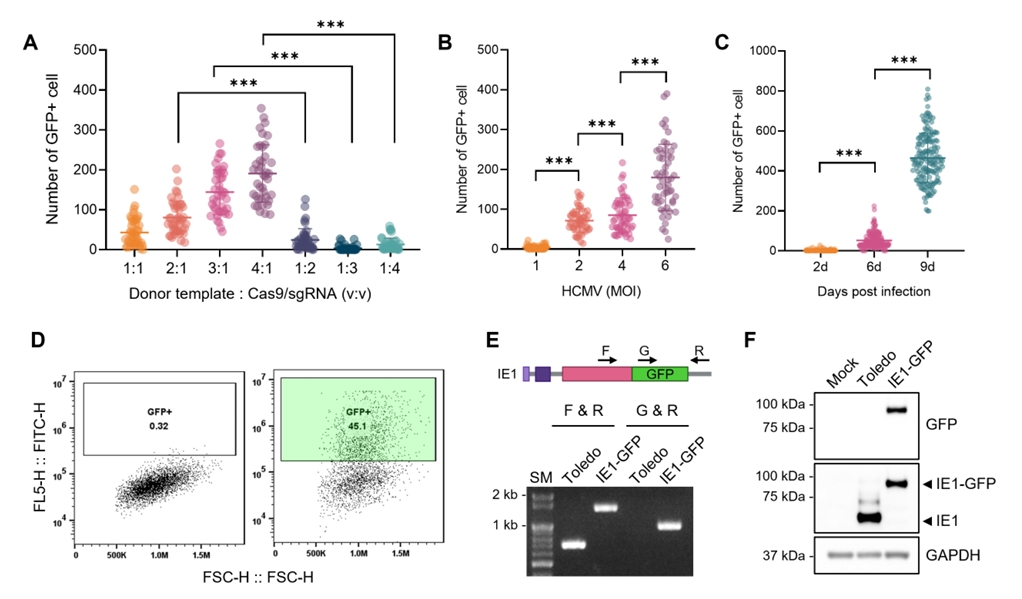
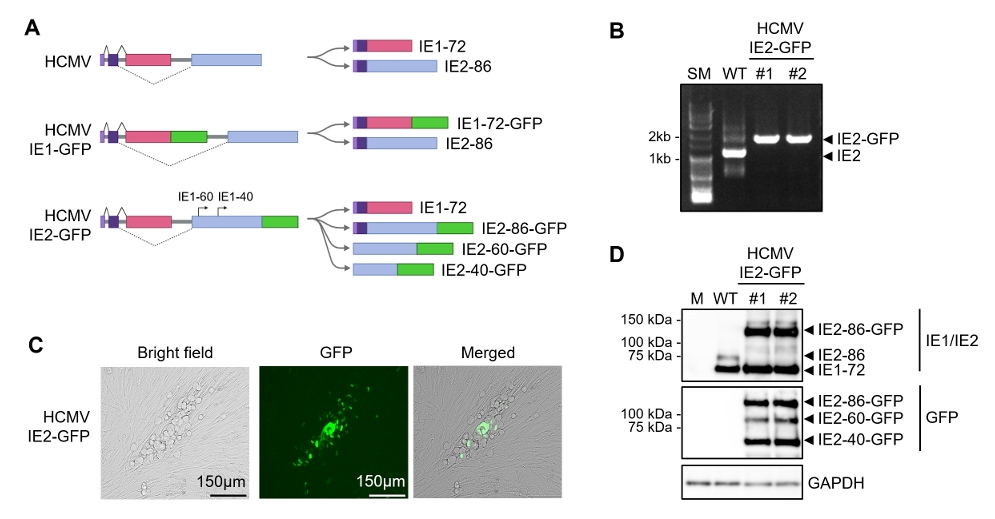
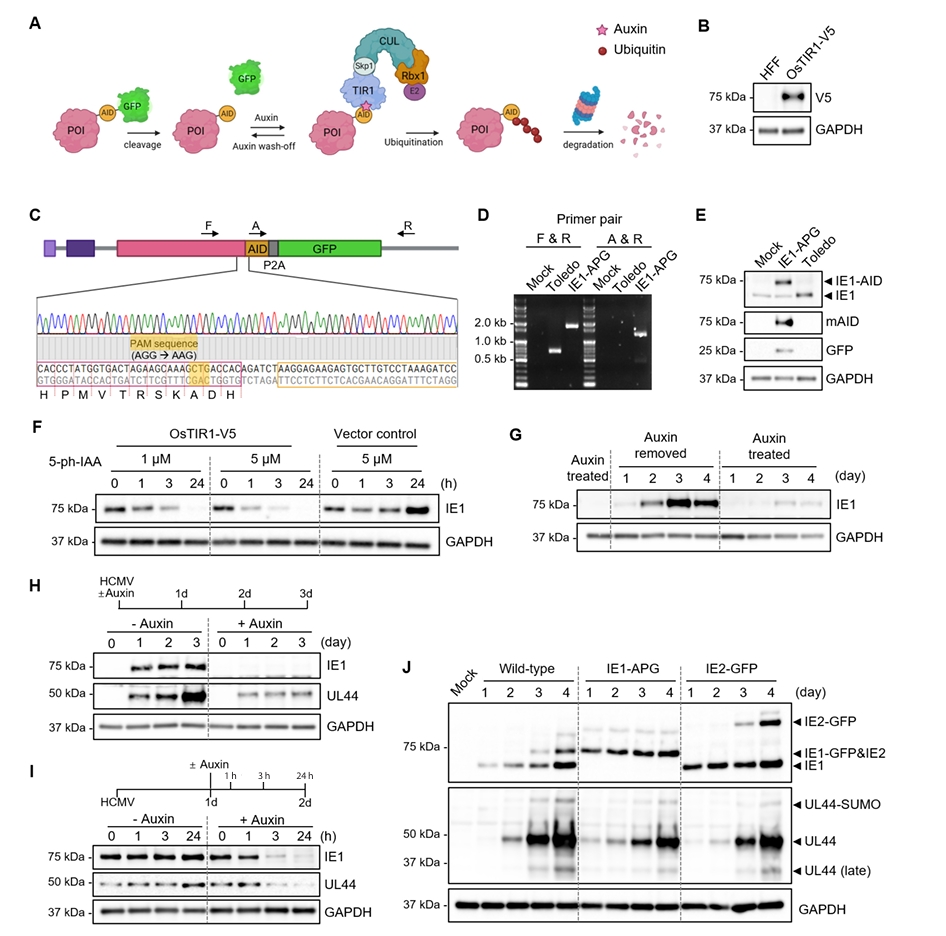






Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
| Name | Sequence (5'-3') |
|---|---|
| IE1 sgRNA F | CACCGCCCTATGGTGACTAGAAGCA |
| IE1 sgRNA R | AAACTGCTTCTAGTCACCATAGGGC |
| IE1 HAS1 F for IE1-GFP | GAAGCAAAGCTGACCACAGATCTATGGTGAGCAAGGGCGAG |
| IE1 HAS1 R for IE1-GFP | CATAACAGTAACTGATATATATACAATAGTTTACTTGTACAGCTCGTCCATGC |
| IE1 HAS2 F for IE1-GFP | AGATCTGTGGTCAGCTTTGCTTC |
| IE1 HAS2 R for IE1-GFP | TAAACTATTGTATATATATCAGTTACTGTTATGGATCC |
| IE1 HAS1 F for IE1-APG | CCTTGCTTCTAGTCACCATAGGGGCAGAACATGTATGAGAACTACATTG |
| IE1 HAS1 R for IE1-APG | GGTGGTGGGTTCCTCAGCACC |
| IE1 HAS2 F for IE1-APG | TAAATAAACTATTGTATATATATCAG |
| IE1 HAS2 R for IE1-APG | CCTTGCTTCTAGTCACCATAGGGTTCATGATATTGCGCACCTTCTC |
| pLenti vector R w/ IE1 cut site | CCCTATGGTGACTAGAAGCAAGGGGAACTCCCAAGCTTATCG |
| pLenti vector F w/ IE1 cut site | CCCTATGGTGACTAGAAGCAAGGTACCGGTTAGTAATGATCGACAATC |
| IE1 PAM sequence mutagenesis F | CCTATGGTGACTAGAAGCAAAGCTGACCACAGATCTAA |
| IE1 PAM sequence mutagenesis R | TTAGATCTGTGGTCAGCTTTGCTTCTAGTCACCATAGG |
| D64V integrase mutagenesis F | ATATGGCAGCTAGTTTGTACACATTTAGAAGGA |
| D64V integrase mutagenesis R | TCCTTCTAAATGTGTACAAACTAGCTGCCATAT |
| IE2 sgRNA F | CACCGAGTCTCAGTAAGTGAAAAAC |
| IE2 sgRNA R | AAACGTTTTTCACTTACTGAGACTC |
| pLenti vector F w/ IE2 cut site | AGTCTCAGTAAGTGAAAAACTGGTACCGGTTAGTAATGATCGACAATC |
| pLenti vector R w/ IE2 cut site | AGTCTCAGTAAGTGAAAAACTGGGGAACTCCCAAGCTTATCG |
| IE2 HAS1 F for IE2-GFP | CCAGTTTTTCACTTACTGAGACTACACCCGCAATCATGAGG |
| IE2 HAS1 R for IE2-GFP | AGATCTCTGAGACTTGTTCCTCAG |
| IE2 HAS2 F for IE2-GFP | TAAGTGAAAAACTGGAAAGAGAGACATGG |
| IE2 HAS2 R for IE2-GFP | CCAGTTTTTCACTTACTGAGACTAGACGGCGTATAGGAGTCC |
| GFP F for IE2 and IE2-GFP | GAGGAACAAGTCTCAGAGATCTATGGTGAGCAAGGGCGAG |
| GFP R for IE2 and IE2-GFP | CTCTTTCCAGTTTTTCACTTACTTGTACAGCTCGTCCATGC |
| WPRE qPCR F | GGCTGTTGGGCACTGACAAT |
| WPRE qPCR R | CCGAAGGGACGTAGCAGAAG |
| TIR1(F74G) F | GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGACATACTTTCCTGAAGAGG |
| TIR1(F74G) R | GGGGACCACTTTGTACAAGAAAGCTGGGTACAGAATCTTCACAAAGTTGGGA |
| IE1 genotyping F | CATGAAGGTCTTTGCCCAGT |
| IE1 genotyping R | CTGCTAACGCTGCAAGAGTG |
| mAID2 F | AGATCTAAGGAGAAGAGTGCTTGTCC |
Table 1.
TOP

