ABSTRACT
- Yeast prion [PSI+], an amyloid form of the translation termination factor Sup35p/eRF3, causes translational stop codon readthrough by sequestering functional Sup35p. This unique phenotype may be analyzed via [PSI+]−suppressible nonsense alleles, and has greatly contributed to the advancement in yeast prion research. For comparing canonical reporters, like chromosomal ade1−14 or ade2−1, and plasmid-borne ura3−14, the de novo generation and characteristics of [PSI+] was investigated across common yeast laboratory strains (BY4741, 74D−694, and 779−6A). The results showed significant variability in [PSI+] induction frequency among strains. [PSI+] was successfully induced in BY4741 and frequently in 74D−694 (via Ade+ selection), but not in 779−6A. Notably, [PSI+] clones, even from identical genetic backgrounds, displayed vastly different nonsense suppression phenotypes depending on the reporter allele used; resulting in diverse growth patterns and suppression levels. Quantitative analyses revealed that prion seed counts fluctuated significantly based on the detection allele and observed phenotype. Furthermore, Sup35p aggregate visualization revealed distinct structural patterns between BY4741 and 74D−694, indicating strain-specific differences. Transferring [PIN+] prion variants from different strains into a common [psi−][pin−] background yielded similar [PSI+] inducibility and seed numbers, suggesting that the observed phenotypic and quantitative diversities of [PSI+] prions stem primarily from the interplay between the specific reporter detection system and the host strain's genetic background rather than solely from inherent differences in the initial [PIN+] prion or fundamental changes in the [PSI+] protein itself. This study underscores the crucial need to consider both the detection methodology and host genetic context for accurate prion variant characterization.
-
Keywords: yeast prion [PSI+], nonsense-suppression
Introduction
Prions are proteinaceous infection particles that were considered mammalian and human pathogens for a long time after the first experimental evidence of their existence (Prusiner, 1982). However, since the discovery of the prions [PSI+] and [URE3] in the model microorganism Saccharomyces cerevisiae, several more prions were reported in budding yeast, and prions were searched in other microbes, such as the fission yeast Schizosaccharomyces pombe, the filamentous fungus Podospora anserina, and even bacteria (Alberti et al., 2009; Coustou et al., 1997; Derkatch et al., 2001; Fleming et al., 2019; Sharma et al., 2024; Son et al., 2023; Sondheimer and Lindquist, 2000; Wickner, 1994; Yuan and Hochschild, 2017). Along with these discoveries, the ease of manipulation of yeast and fungal genetics has led to pioneering studies on prions and host physiology at the cellular level (Son, 2024, 2025).
Yeast [PSI+] is the prion form of the translation release factor Sup35p/eRF3. This factor interacts with Sup45p/eRF1, which is required for stop codon recognition, and. together, they play a role in translation termination (Stansfield et al., 1995). The structural conversion of Sup35p into the [PSI+] prion form results in the inactivation of its own function; thus, translation termination does not occur, neither by readthrough of the termination codon or premature stop codon (nonsense) suppression. Based on the [PSI+] prion phenotype, nonsense-containing alleles have been introduced into yeast strains to detect or score [PSI+] prions. Combinations of SUQ5 (a weak tRNA suppressor) with alleles ade2−1 or ade1−14 (with UAA and UGA stop codons inserted in their ORFs, respectively) have been used as [PSI+] prion-reporter systems (Chernoff et al., 1995; Cox, 1965; Inge-Vechtomov et al., 1988; Liebman et al., 1975). For example, in these ade nonsense allele systems, cells with [PSI+] show a typical white color on rich media (YPD) or in media with limited adenine; they are even able to grow in media without supplementary adenine. In contrast, cells without the prion, i.e., [psi−], are red colored on rich media and not able to grow on media lacking adenine. In addition to chromosomally integrated nonsense alleles, the [PSI+]−suppressible nonsense URA3 gene, inserted into a plasmid, has been used to screen strains with an ura3 mutation and without ade2−1 or ade1−14 mutations, such as S288C and its derivatives (Manogaran et al., 2006). These [PSI+]−suppressible nonsense alleles not only facilitate in−depth studies about the [PSI+] prion by allowing the selection of [PSI+] or [psi−] cells but also provide a plasmid-based [PSI+] scoring system that may be used in a wide range of yeast strains (Son, 2025).
A unique biological property among human, mammalian, and yeast prions is that a single prion protein sequence can give rise to multiple distinct “prion variants” or “strains” (Bradley et al., 2002; Derkatch et al., 1996; King, 2001; Schlumpberger et al., 2001; Tanaka et al., 2004). These prion variants are usually stably propagated, with amyloid filaments templating new monomers to adopt the same conformation as that of the existing amyloid molecules when they join both ends of the filaments (Wickner et al., 2015). In yeast, prion variants were first classified in accordance with the strength of cell phenotypes as “strong” and “weak,” referring to nonsense suppression efficiency of individual [PSI+] isolates or as “lethal” and “mild,” when referring to the degree to which these prions inhibit cell growth or affect cell viability, respectively (Derkatch et al., 1996; Nakayashiki et al., 2005). Later, numerous prion variants were reported to differ in their propagation stability: the degree of transmission from mother to daughter cells during mitosis; chaperone dependence: the ability to propagate in the presence (usually overproduction) or absence of specific chaperones, such as Hsp104; and transmission barrier crossing ability: the capacity to transmit across inter- or intra-species barriers, that is, between different species or within the same species but with different genetic backgrounds, respectively (Wickner et al., 2019).
A cumulus of observations and experimental evidence about prion variants originated the “prion cloud” model, which proposes that a “cloud” of prion variants segregate and mutate at a certain frequency as cells grow. The model signifies the dynamic and heterogeneous nature of prion populations within a cell, suggesting that prions are not static entities, but rather evolving populations, emphasizing the need for prion-handling systems (described below) to be sufficiently robust to handle this heterogeneity (Bateman and Wickner, 2012, 2013). This genetics-based prion variant concept is supported by extensive solid-state NMR studies. These structural approaches have confirmed that yeast prion amyloids (Sup35p, Ure2p, and Rnq1p) have a folded, parallel, in−register β-sheet architecture (Baxa et al., 2007; Shewmaker et al., 2006; Wickner et al., 2008). These resolved structures provide not only an explanation on how these proteins can template their own structure/conformation, as identical residues can favorably interact in this arrangement, ensuring faithful propagation of conformational information, but also an explanation on how a single protein can lead to multiple distinct “prion variants” with stable propagation ability (Tycko, 2014; Wickner et al., 2015).
Most prion variants are pathogenic and severely hinder growth or even kill host cells (Nakayashiki et al., 2005). These pathogenic variants are rarely found in wild strains, suggesting that prions have detrimental effects on cells, and that cells have evolved robust anti-prion systems to counteract them. A series of studies has revealed that yeast has multi-layered anti-prion systems that inhibit the emergence of prion variants, cure those that have already been developed, and mitigate their pathogenicity (Son et al., 2023; Son and Wickner, 2022). For instance, Hsp104 is essential for most yeast prion propagation, but also functions as an anti-prion system by curing many [PSI+] variants arising in its absence (Gorkovskiy et al., 2017). Btn2p and Cur1p are also known to cure most newly formed [URE3] prion variants in btn2Δcur1Δ strains by restoring these proteins (Kryndushkin et al., 2008; Wickner et al., 2014). Nonsense-mediated mRNA decay (NMD) factors (Upf1, 2, and 3p), required for the degradation of aberrant mRNA, can cure newly formed prion variants, possibly by directly binding to prion-forming Sup35p monomers or interfering with fibril elongation (Son and Wickner, 2018). Additionally, Siw14p, which is involved in inositol pyrophosphate metabolism, can cure [PSI+] prion variants (e.g., [PSI+]) by modulating the levels of specific inositol polyphosphate signaling molecules that influence prion dynamics (Wickner et al., 2017). These NMD factors and Siw14p were found in a yeast genetics-based screening, and the combined results suggested the dual functions of each anti-prion component: mRNA quality control of NMD factors or metabolic regulation of Siw14p, respectively, while they both have the ability to counteract [PSI+] prions derived from their own functions.
The Ribosome−associated complex (RAC), consisting of Ssb1/2p (Hsp70)-Zuo1p (Hsp40)-Ssz1p (Hsp70), was previously known for preventing misfolding of newly synthesized polypeptides; the lack of any of them leads to increased [PSI+] prion frequencies (Amor et al., 2015; Chernoff et al., 1999; Gautschi et al., 2001, 2002; Kiktev et al., 2015; Pfund et al., 1998; Rakwalska and Rospert, 2004; Yan et al., 1998). Moreover, most [PSI+] prion isolates formed in RAC component-deletion mutants were cured after the restoration of each of the components, even when expressed at normal levels (Son and Wickner, 2020). Although the previously mentioned anti-prion systems are in charge of prion variants arising in the absence of each of such systems, most [PSI+] prion variants arising in anti-prion system multiple deletion mutants (i.e., ssz1Δupf1Δhsp104T160M strain, suh) were cured through the cooperation of the systems (Son and Wickner, 2022). Taken together, prion variants can be categorized based on their sensitivity to anti-prion systems, with most prion isolates being sensitive to multiple systems.
In addition, a series of studies on [PSI+] prion variants categorized by their propagation diversity have been conducted using well-established prion variant typing (Huang and King, 2020; Huang et al., 2021; King, 2025; Yu and King, 2019). Twenty-three [PSI+] prion variants of WT Sup35p with different propagation abilities (19 novel variants and 4 previously documented variants: VH, VK, VL, and W8) were obtained and confirmed to be unique and not a composite of other variants (Huang and King, 2020). Although a recent study on the [PSI+] prion variants arising in a suh triple mutant of the 74D−694 yeast strain showed contrary results to those of previous reports, it facilitated an in-depth alternative analysis on how ssz1Δ and upf1Δ mutations can independently contribute to or facilitate prion propagation. This study also showed that [PSI+] prion variants are stringently dependent on the appropriate activities of Hsp104 disaggregase, and that these variants can be categorized based on their sensitivity to Hsp104 activity levels (King, 2025). Understanding these yeast prion variants has significant implications for research on human amyloidoses, such as Alzheimer's and Parkinson's diseases. However, yeast prion variants are generally isolated using specific detection systems, such as the nonsense allele for [PSI+], and the yeast strains preferred by each research group. Therefore, a comparative analysis of yeast prion variants isolated through different detection systems and from different strain backgrounds is needed to connect all those previously conducted studies. In this study, we used three representative conventional laboratory yeast strains (BY4741, 74D−694, and 779−6A), widely used for [PSI+] prion studies, and isolated various [PSI+] prion variants with different [PSI+]-suppressible alleles (ade1−14 or ade2−1 and ura3−14). Furthermore, we showed that almost none of the [PSI+] variants isolated using one detection system overlap with those obtained using other systems in their nonsense suppression phenotypes.
Materials and Methods
Nomenclature
Yeast prions are shown in brackets to indicate they are non-chromosomal genes, e.g., [PSI+] and [PIN+]. Phenotypes of yeast strains were marked with superscripted +/−. Dropout (DO) media with corresponding amino acid(s) was designated with − and three letter codes.
Strains, plasmid, and media
This study utilized yeast strains and plasmids detailed in Table 1. All media preparations followed previously established protocols (Sherman, 1991). To induce the expression of Sup35p NM under the control of the GAL1 promoter, cells were grown in media containing 2% galactose and 2% raffinose, a method consistent with prior research (Son and Wickner, 2018). The parental strains, including those with a BY4741 background that carry the ade1−14 allele, were derivatives from earlier studies (Son and Wickner, 2018). For scoring [PSI+] using the suppressible ura3−14 mutation and for inducing prion formation, the pM6/p1520 plasmid was routinely employed. This plasmid is a centromeric vector containing the LEU2 marker, the ura3−14 allele, and the GAL1 promoter driving SUP35 NM expression. To ensure the consistent presence of the pM6 plasmid throughout all [PSI+] scoring experiments, strains and colonies were maintained on media lacking leucine.
[PSI+] seed measurement
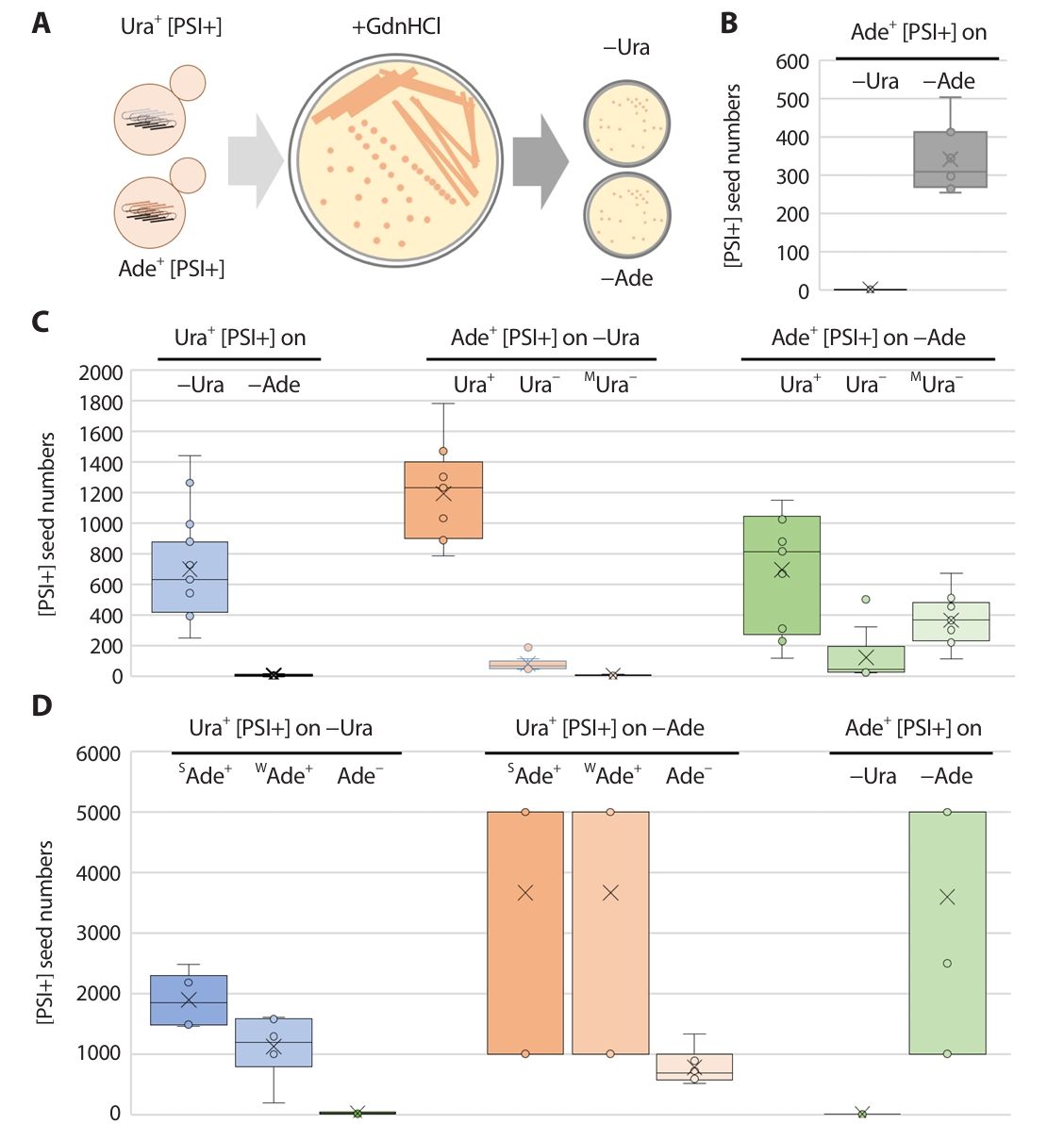
Following the method described by Cox et al. (2003), freshly grown [PSI+] cells were streaked for single colonies on YPAD medium supplemented with 5 mM Guanidine Hydrochloride (GdnHCl). Individual colonies, along with the underlying agar block, were then suspended in distilled water and plated onto media lacking either uracil or adenine. Colonies that exhibited the Ura+ or Ade+ phenotype were counted, with each such colony presumed to represent a [PSI+] prion "seed" present in the cell that initiated that colony (Fig. 1A). For each yeast strain tested, at least ten individual colonies underwent this analysis. A subset of the Ura+ or Ade+ colonies obtained were subsequently checked for their guanidine curability to confirm that the observed phenotype was indeed due to the [PSI+] prion.
Cytoduction
The kar1 mutation results in a failure of nuclear fusion following cytoplasmic fusion during the yeast mating process. Consequently, the cytoplasms of the resulting "daughter" cells are a mixture of those from the two parental strains. This phenomenon is leveraged in cytoduction, a widely used technique for transferring cytoplasmic genes from a donor strain to a recipient strain.
In this process, the transfer of mitochondrial DNA (ρ), indicated by the ability to grow on glycerol (ρ+), is typically used as a marker for successful cytoplasmic transfer. The donor strain is ρ+ (glycerol−utilizing), while the recipient strain is rendered ρo (lacking mitochondrial DNA) by growth in ethidium bromide-containing media. Both donor (carrying its own [PIN+] prion) and recipient ([pin−], previously cured of prions by GdnHCl treatment) strains carried the pM6 plasmid. For cytoduction, donor (ρ+) and recipient (ρo) cells were mixed in distilled water, with approximately a three-fold excess of donor cells, and spotted onto a YPAD plate to facilitate mating. Following incubation at 30°C, the cell mixture was streaked for single colonies on media designed to select against the donor. After a further three days of incubation at 30°C, individual colonies were replica-plated onto YPG (glycerol−containing media, selective for ρ+ cells), media selective for diploids, and media lacking uracil. Clones that grew on YPG but failed to grow on diploid selection media were identified as cytoductants (i.e., cells that received cytoplasm but not a fused nucleus from the donor). The successful transfer of [PIN+] prions from the donor was additionally verified through microscopic observation and by testing for [PSI+] prion inducibility in the cytoductants.
Results
De novo [PSI+] generation differs among laboratory yeast strains
Sup35p overproduction (either the Sup35p N or NM domains) has been reported to induce [PSI+] prion generation (Derkatch et al., 1996; Wickner, 1994). Therefore, [PSI+] was induced in three representative yeast strains that are widely used for [PSI+] prion studies for further analysis. To compare [PSI+] detectability among [PSI+]−suppressible nonsense alleles, such as ade1−14 (on chromosome of BY4741 derivatives and 74D−694) or ade2−1 (on chromosome of 779−6A), strains were transformed with a centromeric plasmid pM6/p1520 (Table 2; Son and Wickner, 2018, 2020, 2022; Wickner et al., 2017), harboring the nonsense allele ura3−14 (Manogaran et al., 2006), and the [PSI+] prion forming domain (NM) of Sup35p under the control of the galactose-inducible GAL1 promoter. Strains were grown in galactose and raffinose for 2 days, cells were serially diluted and plated on media lacking uracil (−Ura) and adenine (−Ade) to select Ura+ and Ade+ [PSI+] clones. For further analysis, the colonies of each strain were tested for prion curability by transient growth on media containing 5 mM guanidine hydrochloride (GdnHCl) (Jung et al., 2002; Jung and Masison, 2001; Tuite et al., 1981).
The frequency of Ura+ [PSI+] clones in 74D−694 was slightly lower than that in BY4741, whereas Ura+ colonies in 779−6A were not curable by GdnHCl, indicating that the arising colonies were not [PSI+] (Table 3). Besides, Ade+ [PSI+] clones appeared most frequently in 74D−694 strain cells on −Ade plates (Table 3). Ade+ colonies were curable in all cases.
Nonsense suppression phenotypes differ depending on the reporter alleles and strains used
Most [PSI+] clones isolated using a certain nonsense allele stably propagated in their own background. However, they sometimes showed dissimilar propagation phenotypes when a different nonsense allele is used, even in the same background (King, 2025; Son and Wickner, 2018; Yu and King, 2019). To test [PSI+]–suppressible phenotypes using two different nonsense allele-based detection systems, GdnHCl–curable Ura+ or Ade+ [PSI+] clones (Table 3) were streaked on non-selective media and replica plated on selective media (–Ura and –Ade). In the BY4741 background, none of the Ura+ [PSI+] clones grew on media lacking adenine (Ade− phenotype), while Ade+ [PSI+] clones showed distinct groupable phenotypes on media lacking uracil (Ura+: 6/18, Ura−: 6/18, and mixed but mostly Ura−: 6/18; Table 4). In the 74–D694 background (ade1−14), in contrast, Ura+ [PSI+] clones showed groupable growth phenotypes on –Ade plates (Strong Ade+: 3/18, Weak Ade+: 6/13, and Ade−: 4/13). Though most Ade+ [PSI+] clones were Ura−, approximately 20% of the subclones of a fraction of these clones (12/18) showed an unstable Ura+ phenotype (Table 4). In the 779–6A strain background (ade2–1), isolatable Ade+ [PSI+] clones had a uniform phenotype (Table 4). We also tested strains L2892 (with a strong [PSI+] variant) and L2885 (with a weak [PSI+] variant), which were previously isolated using the ade1−14 allele and have been conventionally used in many [PSI+] experiments (Mathur et al., 2009). L2892 showed even frequencies of Ura+ and Ura− phenotypes, whereas L2885 showed a uniform Ura− phenotype on –Ura media (Table 4).
Seed numbers of [PSI+] variants isolated by different detection systems
The number of prion seeds (propagons) or infectious prion particles in each prion-carrying strain was calculated using GdnHCl, which is a specific inhibitor of the Hsp104 chaperone that is required for prion propagation via amyloid filamentation (Cox et al., 2003). Millimolar treatment with GdnHCl halts amyloid filamentation, and new filaments are no longer generated. Therefore, the number of colonies arising on prion-selectable media after the spreading of a single colony on a GdnHCl-containing medium reflects the number of preexisting seeds in a prion-carrying strain (Fig. 1A).
The number of seeds of Ade+ [PSI+] clones in the 779–6A background were approximately 350 on –Ade plates, but 0 on –Ura plates, indicating that the ura3–14 allele could not detect the seeds of the Ade+ [PSI+] clones (Fig. 1B).
In the BY4741 background, Ura+ [PSI+] clones (with a uniform Ade− phenotype) showed seed numbers that ranged between 400 and 900 when detected on –Ura plates, but that were undetectable by the ade1–14 allele. Regarding Ade+ [PSI+] clone seed numbers calculated on –Ura media, only those with a Ura+ phenotype showed reliable seed numbers, which varied between 900 and 1,400 (Fig. 1B). The numbers of Ade+ [PSI+] clones on –Ade media correlated with their Ura+/Ura− phenotypes: 300–1,000 seeds for Ura+ clones, less than 200 seeds for Ura− clones, and 200–500 seeds in mixed but mostly Ura− clones (Fig. 1C).
In the 74–D694 background, Ura+ [PSI+] clone seed numbers were calculated on –Ura media and correlated with their Ade+/Ade− phenotypes, therefore, the seed number increased as the strength of Ade phenotype increased (Fig. 1D). When using the ade1–14 allele (on –Ade media) for detecting Ura+ or Ade+ [PSI+] clones, seed numbers showed wide fluctuations (103 or over 103) and only the seed number of Ura+ [PSI+] clones with an Ade− phenotype could be measured: 600–1,000 seeds. The number of seeds of Ade+ [PSI+] clones on –Ade media showed the same type of fluctuation, and almost no seeds were obtained on –Ura media (Fig. 1D).
Aggregation of [PSI+] prion variants in different strain backgrounds
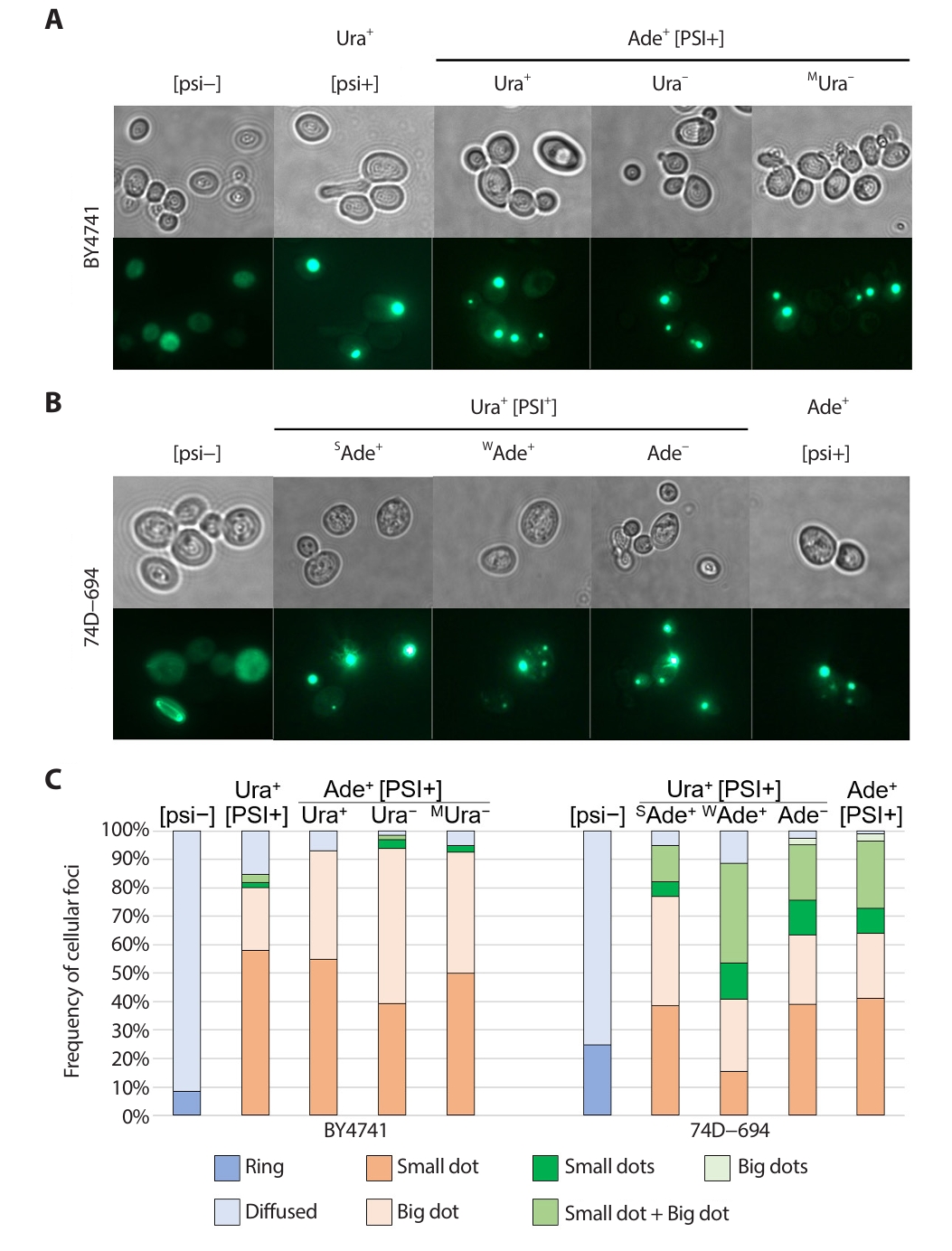
The expression of GFP-tagged prion proteins is used to visualize preexisting [PSI+] prion aggregates in cells (Son and Wickner, 2018, 2020). To investigate whether the discrepancy in prion aggregate formation depended on strain background, isolation method, or phenotype, Sup35 NM-GFP was transiently overexpressed in each strain (Fig. 2). [psi–] strains of both backgrounds consistently showed a diffuse GFP signal (no prion aggregates) and rare formation of a ring structure (a sign of ongoing prion generation). In contrast, [PSI+]-carrying strains faithfully showed GFP foci (Fig. 2A and 2B; Mathur et al., 2010; Son and Wickner, 2018, 2020)
The formation of prion aggregate structures has been reported to be affected by prion variants with different characteristics, but without any chromosomal mutation (wild-type strain) (Huang et al., 2013; Zhou et al., 2001). To investigate the different aggregate formations among strains, the frequency of cells with different aggregates was calculated and sorted by the number or size of the dot(s). Generally, [PSI+]s carried by BY4741 strain backgrounds had single foci with evenly distributed small and large size dots (Fig. 2C). Importantly, no remarkable differences were found among the Ura+ [PSI+] and Ade+ [PSI+] isolates or among Ade+ [PSI+] variants with different ura3–14 nonsense suppression phenotypes. However, [PSI+]s in 74–D694 strain backgrounds showed multiple foci at relatively high frequencies, with a combination of small and large dots (Fig. 2C). Ura+ [PSI+] isolates with a strong Ade+ phenotype had a relatively high frequency of single dots per cell, whereas Ura+ [PSI+] isolates with a weak Ade+ phenotype had a relatively high frequency of multiple dots per cell.
[PSI+] inducibility by [PIN+] from different strain backgrounds
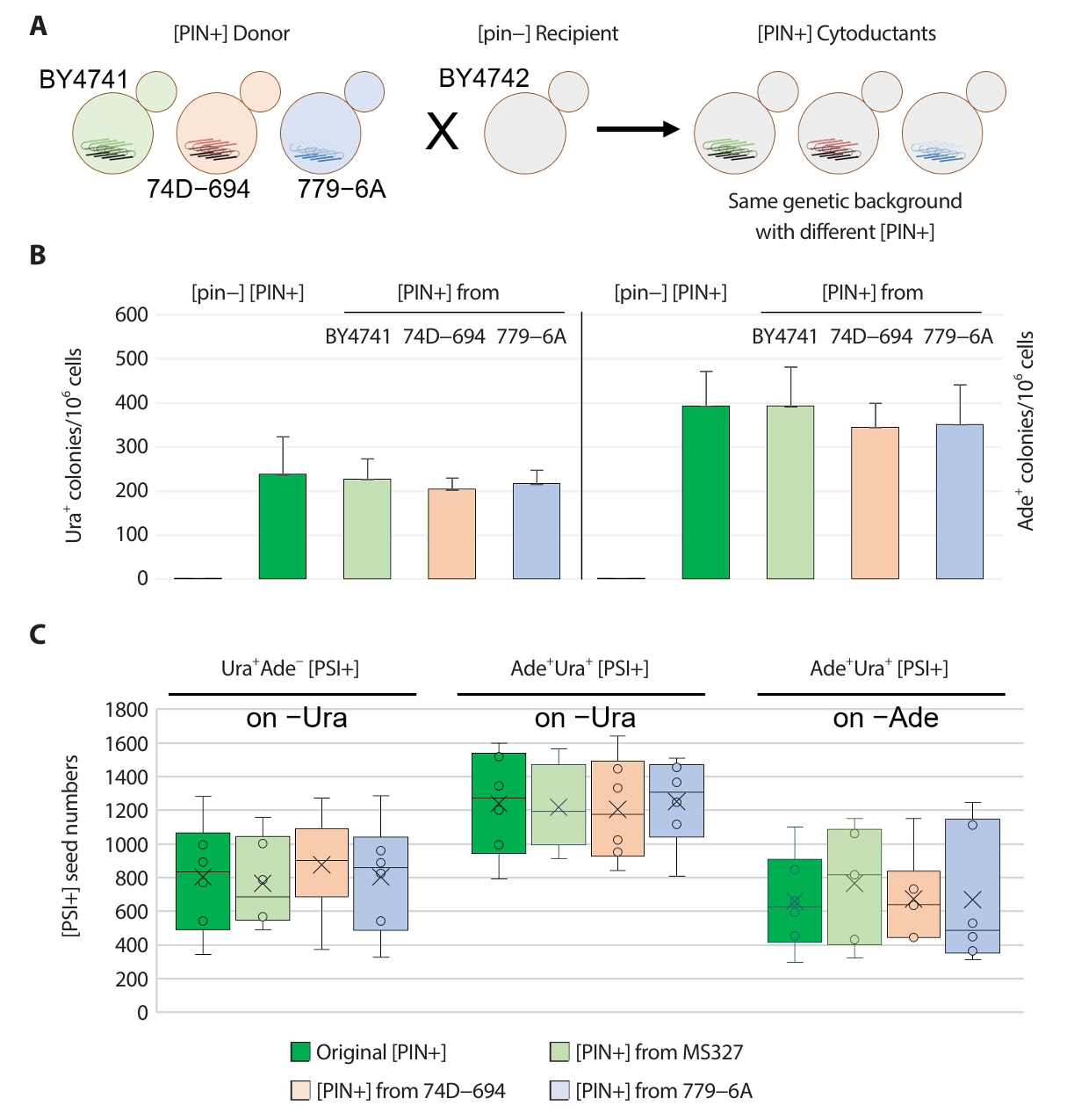
The characteristics of certain prion variants can be altered by genetic alterations such as the deletion or overproduction of prion-related genes, even in isogenic genetic backgrounds, or changes in genetic backgrounds. For example, the deletion of Hsp90 and its cochaperones was reported to alter [PIN+] prions in isogenic strains, leading to a variety of changes, such as alteration in [PSI+] prion induction efficiency, cellular aggregate formation, and genetic dominance of prion variants (Lancaster et al., 2013). To compare the characteristics of [PIN+] prion variants in each strain, [PSI+] inducibility among [PIN+] strains from different backgrounds and the phenotypes of the resulting [PSI+] isolates were analyzed. Each [PIN+] prion variant was transferred into BY4741 [psi−][pin−] WT recipient strain prepared by transient GdnHCl treatment, and [PSI+]s were then induced in such WT strains with different [PIN+] variants from BY4741, 74–D694 and 779–6A strain (Fig. 3A).
The frequency of Ura+ [PSI+] clones induced by overproduced Sup35p NM was approximately 200 per 106 cells, and that of Ade+ [PSI+] clones was approximately 350 per 106 cells (Fig. 3B, Table 3). The nonsense suppression phenotype of Ura+ [PSI+] isolates in WT strains with [PIN+] variants from different sources was consistently Ade– on media lacking adenine (Table 1). Ade+ [PSI+] clones showed different ura3–14 suppression phenotypes depending on the isolates (Ura+: 4–5/12, mostly Ura+: 1–3/12, and Ura–: 4–6/12) (Table 1).
The number of Ura+ and Ade+ [PSI+] clones was determined as described above. Ade– showing Ura+ [PSI+] clones had approximately 500–1,000 seeds per cell on –Ura media, and Ura+ showing Ade+ [PSI+] clones harbored 1,000–1,500 and 400–1,000 seeds per cell when detected on –Ura and –Ade media, respectively (Fig. 3C). These results suggest that all the [PIN+] variants from different strains have the same [PSI+] inducibility, resulting in [PSI+] isolates with very similar seed numbers.
Discussion
In yeast cells without the [PSI+] prion ([psi–] state), Sup35p (eRF3) is soluble, fully functional, and works with Sup45p (eRF1) to efficiently recognize stop codons (UAA, UAG, and UGA) in mRNA, leading to the precise termination of protein synthesis. When Sup35p is converted to the [PSI+] prion form, it misfolds and aggregates into insoluble amyloid fibrils. This sequestration of functional Sup35p leads to a decreased availability of active Sup35p for an efficient translation termination. Therefore, the ribosome often reads through stop codons, allowing translation to continue past where it should normally stop. This is known as nonsense suppression (Paushkin et al., 1997; Stansfield et al., 1995). This nonsense suppression effect by [PSI+] is what makes [PSI+] easily detectable, primarily through nonsense mutation-containing alleles, such as ade1−14, ade2−1, and ura3−14.
In this study, we investigated how de novo [PSI+] prion generation, detection, and characteristics vary across common laboratory yeast strains (BY4741, 74D−694, and 779−6A) using different nonsense suppression alleles. While [PSI+] could be induced in BY4741 and 74D−694 (with higher frequency in 74D−694 via Ade+ selection), the 779−6A strain did not yield true [PSI+] prions that could be detected through Ura+ phenotypes. Notably, [PSI+] clones isolated using one detection system (e.g., ura3−14 for Ura+ selection) often displayed different nonsense suppression phenotypes when tested with another allele (e.g., ade1−14 for Ade+) (Fig. 4), revealing a complex relationship between prion variants, strain backgrounds, and reporter systems. Unexpectedly, we found that the frequency of [PSI+] prion variants generated by Sup35p NM overexpression was higher than that previously obtained using a single detection system. Although the sum of [PSI+]s detected by the two systems may not capture every aspect, overexpression of Sup35p NM induced the generation of more diverse and numerous [PSI+] prion variants than anticipated.
Quantitative analysis of prion seed numbers and aggregate structures further highlighted these discrepancies, showing that seed counts varied widely depending on the detected allele and phenotype, and the aggregate morphology differed between BY4741 (single focus) and 74D−694 (multiple foci) (Fig. 4) backgrounds. Interestingly, despite these phenotypic differences, the underlying [PIN+] prion variants from different strains neither affected [PSI+] inducibility nor the resulting prion seed numbers of [PSI+] variants when transferred to a common [psi−][pin−] background, suggesting that the observed variability primarily stems from the specific detection system and strain context rather than the initial [PIN+] seed itself.
Why do different detection systems lead to different [PSI+] prion variant phenotypes?
Nature of the reporter system and its requirements: In all strains, Ura+ and Ade+ [PSI+] isolates showed significant differences regarding generation frequencies, phenotypes, and propagon number (Fig. 1, Tables 3 and 4). For example, the frequency of appearance of Ura+ [PSI+]s was relatively lower than that of Ade+ [PSI+]s even under the same induction conditions. Furthermore, Ura+ [PSI+]s were not Ade+ at all, but Ade+ [PSI+]s showed complex phenotypes that were even mixed with Ura+ or Ura− phenotypes in certain isolates (Table 4).
The ade1−14 nonsense allele carries a TGG-to-TGA mutation at codon 244 in the ADE1 gene, while the ade2−1 nonsense allele features a GAA-to-TAA mutation at codon 64 in the ADE2 gene, and they both introduce premature stop codons, compared with each of the WT alleles (Cox, 1965; Inge-Vechtomov et al., 1988; Liebman et al., 1975; Roman, 1956). Unlike other characterized mutations on ADE genes, the ura3−14 allele was engineered, and an 81 nucleotide region (codons 234−254) from the ade1−14 allele was inserted between the PGK1 promoter and the URA3 gene by in−frame insertion mutagenesis (Manogaran et al., 2006). These structural differences in nonsense alleles may lead to different mRNA contexts surrounding the stop codon; alternatively, the proteins they encode (ADE1/ADE2 vs. URA3) may have different levels of tolerance to stop codon readthrough (Chernoff et al., 2002; Fitzpatrick et al., 2011; Namy et al., 2003). For example, a partial readthrough that produces just enough functional Ura3 protein to allow growth on −Ura media might not be enough to produce functional Ade1/Ade2 proteins to grow on −Ade media, meaning that the different thresholds of nonsense suppression due to a certain [PSI+] prion variant might induce "strong enough" readthrough for one reporter but not for another.
Proteins encoded by ADE1/ADE2 and URA3 play different roles in the cell. Even if both reporter genes are affected by nonsense suppression, the physiological consequences or the amount of functional protein required for a noticeable phenotype can vary. The specific functions of each of these genes may have different effects on [PSI+] prion detectability of nonsense alleles during the selection process.
The ade alleles used in this study are typically integrated into the chromosome, while the ura3−14 allele is on a plasmid. This might lead to differences in gene dosage or chromosomal context, which can influence expression levels and, consequently, the sensitivity of the detection system. Additionally, despite being designed with a yeast centromeric region (CEN), multiple studies indicate that the actual copy number of CEN plasmids can be “higher than single copy,” and often in the range of three to five copies per cell (Flagg et al., 2019). Thus, the physical location of the chromosome and copy number of pM6 may affect detectability.
Prion variant/strain conformation: The core reason for prion variant diversity is that Sup35p can misfold into many distinct amyloid conformations. Each of these conformations acts as a unique template for further Sup35p aggregation (Frederick et al., 2014; Ohhashi et al., 2018; Toyama et al., 2007). These different Sup35p conformations, even if they all lead to some level of nonsense suppression, might do so with varying efficiencies. For example, the "strong" [PSI+] variant (L2892) might cause a highly efficient readthrough phenotype, affecting multiple nonsense reporters, showing stable Ade+ and evenly distributed Ura+ and Ura− phenotypes. In contrast, the "weak" variant (L2885) might only cause enough readthrough for the most sensitive reporter, showing Ade+ and a uniform Ura− phenotype (Table 4). Moreover, stable Ade+ [PSI+]s in BY4741 can be categorized by their sensitivity to the ura3−14 allele-based reporter as Ura+ (6/18), Ura− (6/18), and mostly Ura− (6/18) (Table 4). Taken together, these results suggest that amyloid conformational diversity might lead to different translational impacts on other detection reporter systems, resulting in different sensitivities.
Strain−specific genetic background (host factors): The stringencies of the three reporter systems were completely different among the strains used in this study. An antithetic operation between BY4741 and 74D−694 was observed, whereas the ura3−14 allele-based reporter system not working at all in 779−6A (Table 3). In addition, subtle differences in Sup35p aggregation were observed between the two strains tested. In the 74D−694 strain, multiple foci were more frequently detected by galactose-induced Sup35 NM GFP expression (Fig. 2). In addition, different strains can exhibit variations in the expression levels or activities of cellular chaperones (such as Hsp104, Hsp70s, and Hsp40s) and other anti-prion factors (such as Btn2p, Cur1p, NMD factors, Siw14p, and RAC components) (Gorkovskiy et al., 2017; King, 2025; Son and Wickner, 2018; Wu et al., 2023). Chaperones are crucial for prion propagation, the fragmentation of aggregates (which create new "seeds"), and even curing. Differences in these host factors can modulate the apparent strength or stability of prion variants, leading to different phenotypic readouts. This may also affect the mechanism of Sup35p aggregation (e.g., the number of aggregates per cell, size, stability, and interaction with chaperones) in different strains. These subtle structural differences can lead to different levels of functional Sup35p sequestration and, thus, different degrees of nonsense suppression in different strains.
BY4741 is a direct descendant of the S288C lineage, the first strain whose genome was fully sequenced. Strains 74D−694 and 779−6A have been extensively used and are particularly prominent in the field of prion research. Direct comparisons of strain genomes have revealed that even closely related laboratory strains can harbor single nucleotide polymorphisms (SNPs) or other genetic variations that might subtly or significantly influence protein folding, quality control, translation, or the stability of prion aggregates (Drozdova et al., 2016; Fitzpatrick et al., 2011). These differences in "modifier genes" could directly alter the phenotypic expression of prion conformations or affect adenine or uracil biosynthesis pathways independently of the nonsense suppression, indirectly influencing the reporter phenotype in different strains.
Closing remarks
In essence, the apparent differences in prion variants among different detection systems are not due to changes in the underlying prion proteins themselves, but rather because: 1) the reporter systems have different sensitivities or functional requirements; 2) specific prion conformations (variants) have inherent differences in their ability to induce nonsense suppression, and 3) the genetic background of a host strain can modify the expression or propagation efficiency of prion variants, thereby altering the observed phenotype. This complexity underscores the challenge and importance of precisely characterizing prion variants, and highlights why researchers often use multiple detection methods and different genetic backgrounds to gain a comprehensive understanding of prion biology.
Acknowledgments
I truly thank Daniel C. Masison, Reed B. Wickner (National Institutes of Health, National Institute of Diabetes and Digestives and Kidney Diseases, Bethesda, MD, USA) and Susan W. Liebman (University of Nevada, Reno, NV, USA) for plasmid and strains. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2022R1A2C1092397) (M.S.).
Fig. 1.The seed number measurement of [PSI+] isolates. (A) Experimental procedure of the measurement of [PSI+] prion seed number. Ura+ or Ade+ [PSI+] isolates in each strain were streaked on 5 mM GdnHCl containing media. Separated single colonies were resuspend and further spread on the media lacking uracil (−Ura) or adenine (−Ade). The number of arising Ura+ or Ade+ single colonies were regarded as pre−existing [PSI+] prion seeds in each strain. (B) The seed number of Ade+ [PSI+] isolates in 779−6A were determined (n = 12). (C) The seed numbers of [PSI+]s in BY4741 were counted (n = 12). Ura+ [PSI+] isolates were determined on different media (light blue, left panel). Ade+ [PSI+] isolates with three different uracil auxotrophic phenotypes (Ura+, Ura−, and MUra−) seed numbers were confirmed on the media lacking uracil (−Ura, light orange, middle panel) or adenine (−Ade, light green, right panel). (D) The seed numbers of [PSI+]s in 74D−694 were counted (n = 12). Ura+ [PSI+] isolates with three different adenine auxotrophic phenotypes (SAde+, WAde+, and Ade−) seed numbers were confirmed on the media lacking uracil (−Ura, light blue, left panel) or adenine (−Ade, light orange, middle panel). Ade+ [PSI+] isolates were determined on different media (light green, right panel). Arising Ura+ or Ade+ ([PSI+]) clones were tested for GdnHCl curability using transient growth on plates containing 5 mM guanidine.
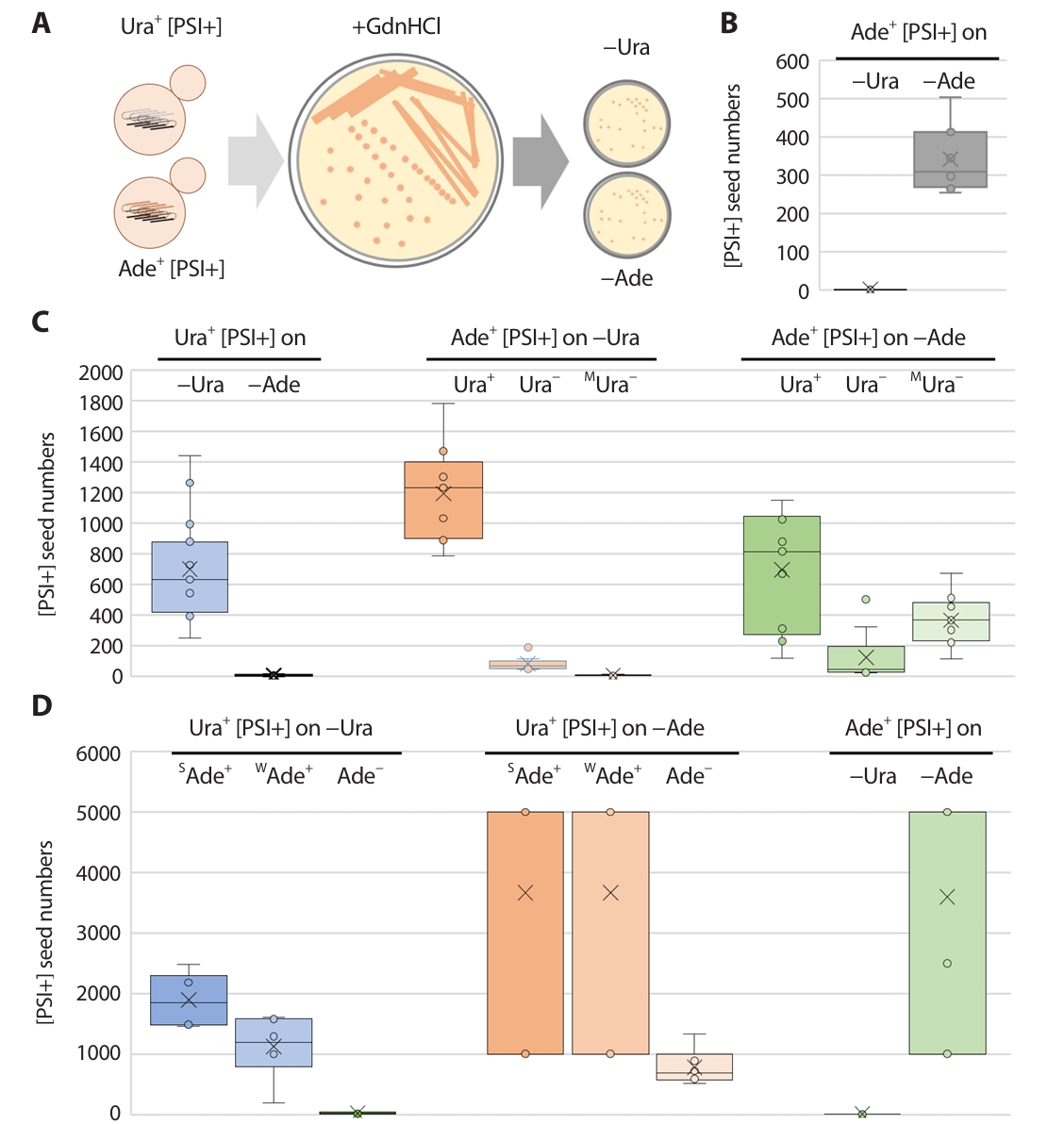
Fig. 2.Sup35 NM−GFP aggregates in [PSI+] isolates. [psi−] strain and original Ura+ or Ade+ [PSI+] isolates with different phenotypes (left to right) were transformed with a CEN plasmid pM107 encoding Sup35 NM-GFP controlled by the ADH1 promoter. The original isolates and their phenotypes examined is shown above the image. After Incubation for at least 16 h in 2% (wt/vol) glucose minimal medium, Sup35 NM-GFP aggregates or diffused GFP signal were observed using fluorescence microscopy (Imager M2, Zeiss). (A) Sup35 NM−GFP aggregates in BY4741 strain background. (B) Sup35 NM−GFP aggregates in 74D−694 strain background. (C) The frequencies of cells with different foci in BY4741 and 74D−694 strain were determined. The frequencies of cells with aggregates were scored in 100−300 cells. [psi−] strain and the original isolates and their phenotypes examined is shown above the bar.
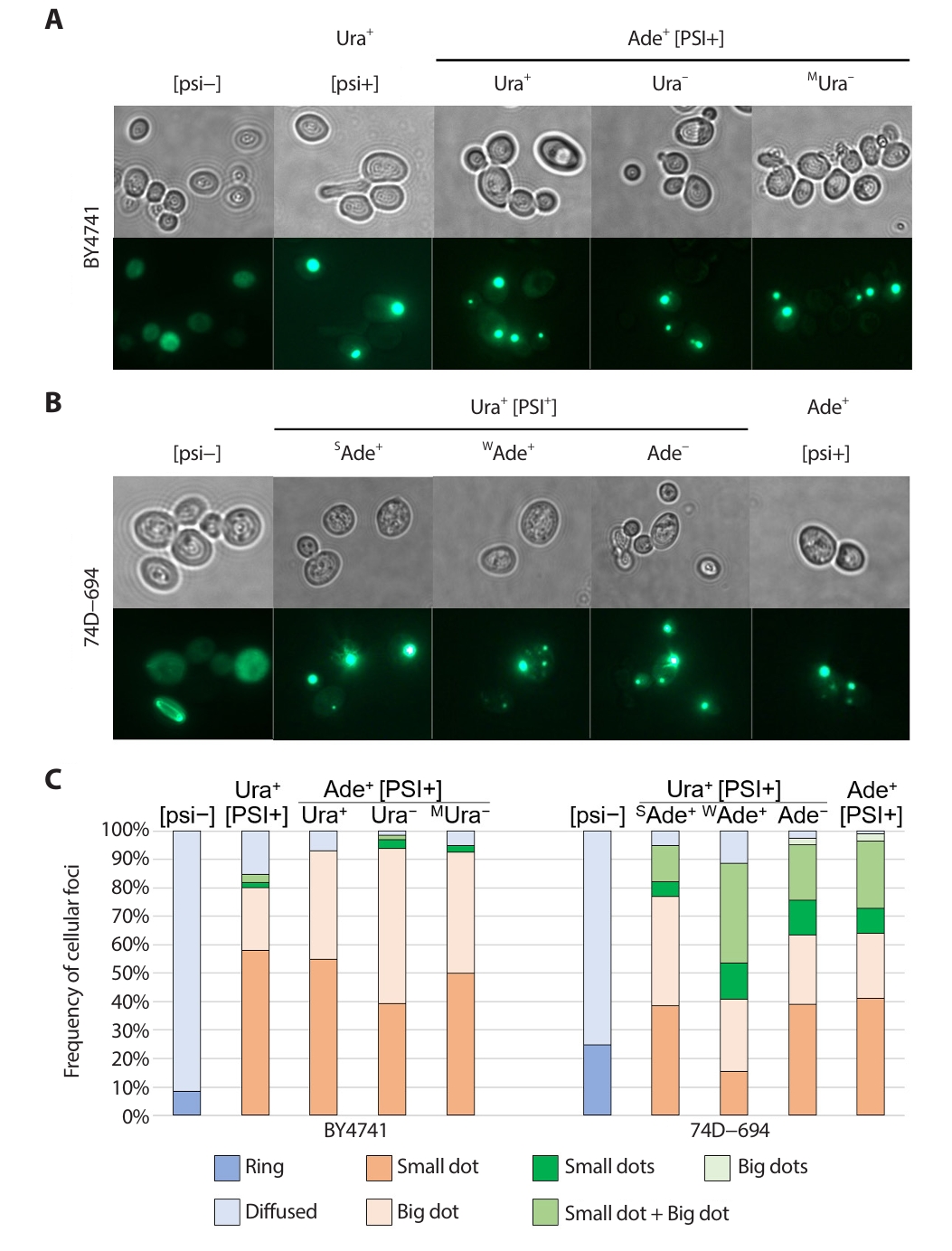
Fig. 3.Comparative analysis of [PIN+] prion introduced from BY4741, 74D−694 and 779−6A strain. (A) All the [PIN+]s from different sources was introduced by cytoduction into [pin−] strain (BY4742/MS173). Then the [PIN+] in stains was transferred back into MS327 [pin−] strain (previously made [pin−] by GdnHCl) by cytoduction. [PSI+] was generated by overproduction of Sup35 NM in 2% (wt/vol) raffinose, 2% (wt/vol) galactose minimal medium. Cells were plated on −Ura or −Ade plates, and arising Ura+ and Ade+ ([PSI+]) clones were tested for GdnHCl curability using transient growth on plates containing 5 mM guanidine. (B) The efficiency of [PSI+] inducibility of [PIN+]s. MS327 [pin−] strain, original [PIN+] carrying strain and [PIN+] from corresponding strain were shown on the bar (left to right). (C) The seed numbers of [PSI+]s isolated in strain carried with different sources of [PIN+]s (n = 12). The seed numbers of Ura+ or Ade+ [PSI+] isolates with different auxotrophic phenotypes confirmed by other reporter system were confirmed on −Ura media and −Ade media. Arising Ura+ or Ade+ ([PSI+]) clones were tested for GdnHCl curability using transient growth on plates containing 5 mM guanidine.
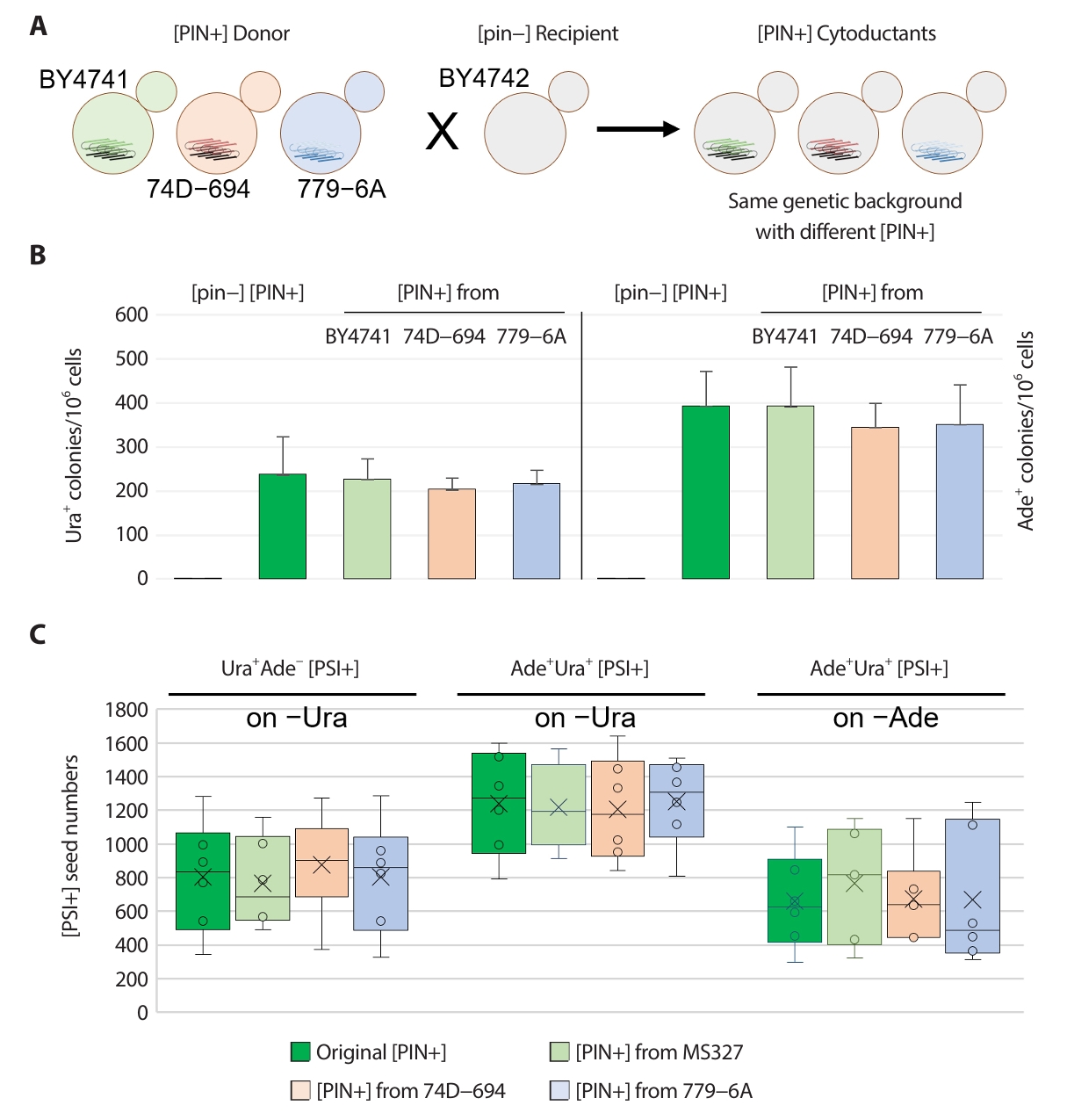
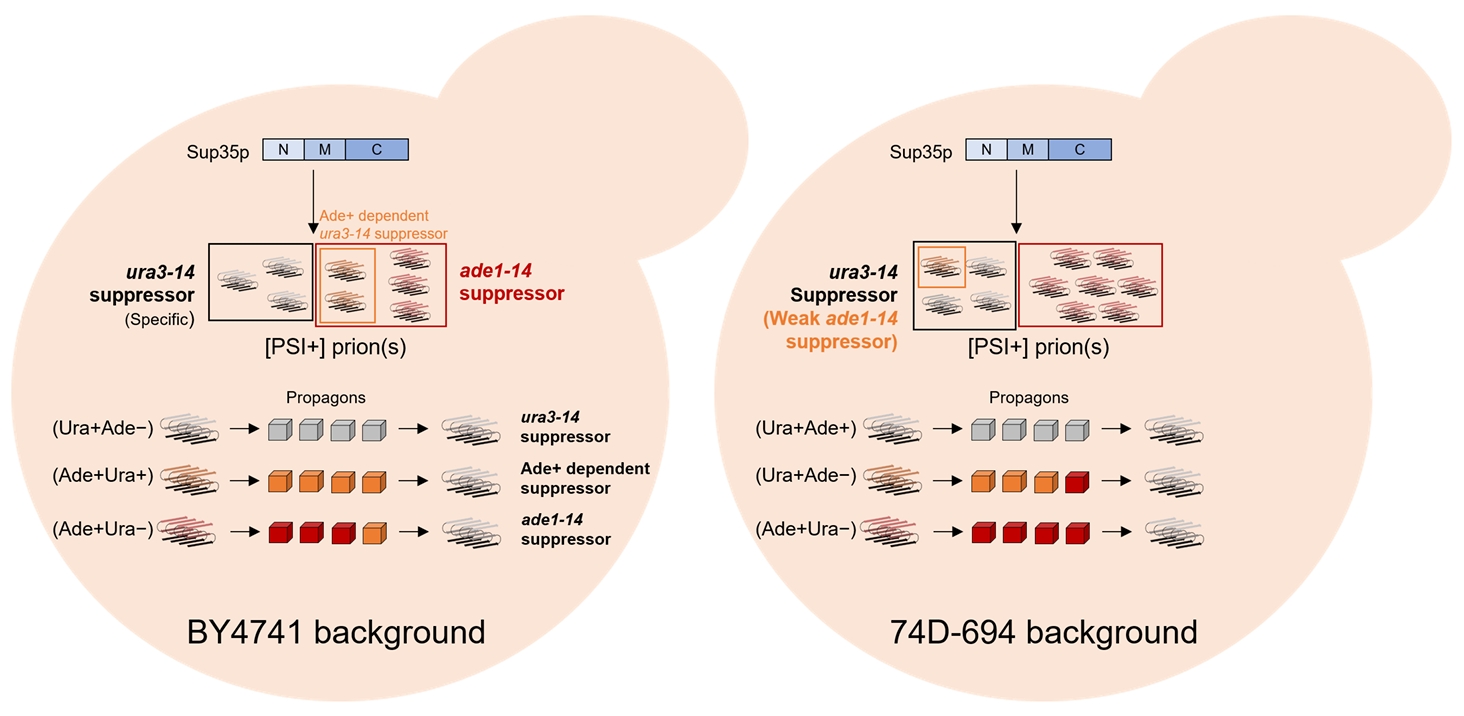
Fig. 4.Detection system− and strain− dependent diversity of yeast [PSI+] prion. During the conversion of Sup35p to [PSI+] prion(s), prion amyloid with different characteristics were appeared. In BY4741 strain background (Left), prion amyloids with grey and red color are able to suppress ura3−14 and ade1−14 in a specific manner, respectively, while amyloid of orange color can be detected by both systems. Cubes with different colors designate the prion propagon from amyloid of same color and are showing same phenotype with each amyloid. In 74D−694 strain, the detectability appears in reverse: ura3−14 allele is generic detection system but ade1−14 allele is specific detection system.
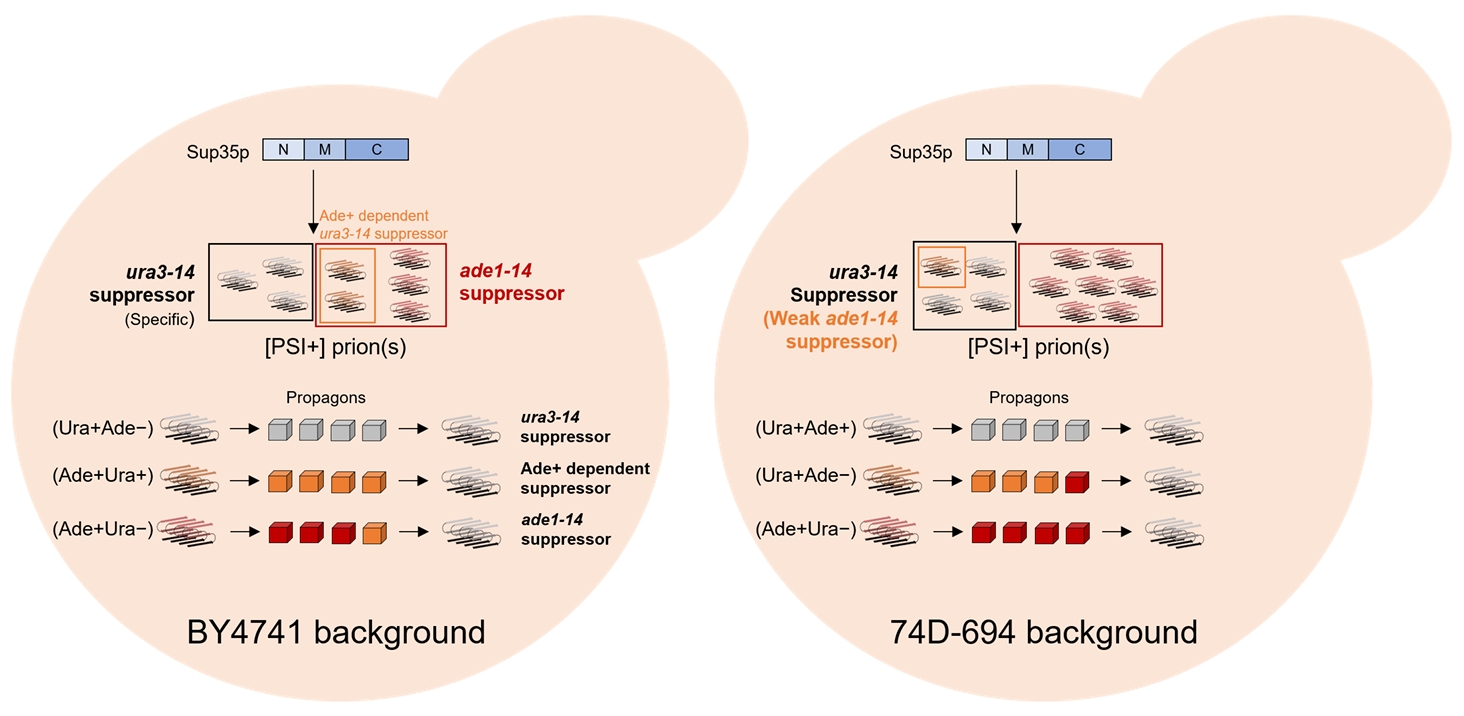
Table 1.Strains and plasmids used in this study
Table 2.The frequency of de novo [PSI+] generation in BY4741, 74D−694, and 779−6A strain
|
Strain |
Total Ura+ colonies per 106 cells |
Total Ade+ colonies per 106 cells |
|
BY4741 |
191.7 ± 39.6 |
372.3 ± 69.6 |
|
74D−694 |
135.3 ± 51.3 |
978.6 ± 84.8 |
|
779−6A |
*17.3 ± 3.5 |
51.0 ± 11.1 |
Table 3.The nonsense−suppression phenotype of [PSI+] isolates in BY4741, 74D−694, and 779−6A strain
|
Strain |
Ura+ [PSI+] isolates on −Ade plate |
Ade+ [PSI+] isolates on −Ura plate |
|
BY4741 |
Uniformly Ade− (18/18) |
Ura+ (6/18) |
|
Ura− (6/18) |
|
aMostly Ura− (6/18) |
|
74D−694 |
bStrongly Ade+ (3/13) |
Ura+ (0/18) |
|
cWeakly Ade+ (6/13) |
Ura− (6/18) |
|
Ade− (4/13) |
dMostly Ura− (12/18) |
|
eL2892 |
N/A |
Evenly Ura+ and Ura−
|
|
fL2885 |
N/A |
Ura−
|
|
779−6A |
gN/A |
*Uniformly Ura− (18/18) |
Table 4.The nonsense−suppression phenotype of [PSI+] isolates in BY4741with different [PIN+] from BY4741, 74D−694, and 779−6A strain
|
Source of [PIN+] |
Ura+ [PSI+] isolates on −Ade plate |
Ade+ [PSI+] isolates on −Ura plate |
|
original |
Uniformly Ade− (10/10) |
Ura+ (5/12) |
|
Ura− (5/12) |
|
aMostly Ura− (2/12) |
|
BY4741 |
Uniformly Ade− (12/12) |
Ura+ (4/12) |
|
Ura− (5/12) |
|
aMostly Ura− (3/12) |
|
74D−694 |
Uniformly Ade− (12/12) |
Ura+ (4/11) |
|
Ura− (6/11) |
|
dMostly Ura− (1/11) |
|
779−6A |
Uniformly Ade− (12/12) |
Ura+ (4/11) |
|
Ura− 4/11) |
|
dMostly Ura− (3/11) |
References
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 137: 146–158. ArticlePubMedPMC
- Amor AJ, Castanzo DT, Delany SP, Selechnik DM, van Ooy A, et al. 2015. The ribosome-associated complex antagonizes prion formation in yeast. Prion. 9: 144–164. ArticlePubMedPMC
- Bateman DA, Wickner RB. 2012. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: intraspecies 'species barriers'. Genetics. 190: 569–579. ArticlePubMedPMCPDF
- Bateman DA, Wickner RB. 2013. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 9: e1003257. ArticlePubMedPMC
- Baxa U, Wickner RB, Steven AC, Anderson D, Marekov L, et al. 2007. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry. 46: 13149–13162. ArticlePubMed
- Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. 2002. Interactions among prions and prion "strains" in yeast. Proc Natl Acad Sci USA. 99: 16392–16399. ArticlePubMedPMC
- Chernoff YO, Lindquist SL, Ono B-I, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science. 268: 880–884. ArticlePubMed
- Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI] prion. Mol Cell Biol. 19: 8103–8112. ArticlePubMedPMCLink
- Chernoff YO, Uptain SM, Lindquist S. 2002. Analysis of prion factors in yeast. Methods Enzymol. 351: 499–538. ArticlePubMed
- Coustou V, Deleu C, Saupe S, Begueret J. 1997. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 94: 9773–9778. ArticlePubMedPMC
- Cox BS. 1965. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity. 20: 505–521. ArticlePDF
- Cox BS, Ness F, Tuite MF. 2003. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics. 165: 23–33. ArticlePubMedPMCPDF
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. 2001. Prions affect the appearance of other prions: the story of [PIN+]. Cell. 106: 171–182. ArticlePubMed
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 144: 1375–1386. ArticlePubMedPMCPDF
- Drozdova PB, Tarasov OV, Matveenko AG, Radchenko EA, Sopova JV, et al. 2016. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strains of the Peterhof genetic collection. PLoS One. 11: e0154722. ArticlePubMedPMC
- Fitzpatrick DA, O'Brien J, Moran C, Hasin N, Kenny E, et al. 2011. Assessment of inactivating stop codon mutations in forty Saccharomyces cerevisiae strains: implications for [PSI+] prion-mediated phenotypes. PLoS One. 6: e28684. ArticlePubMedPMC
- Flagg MP, Kao A, Hampton RY. 2019. Integrating after CEN Excision (ICE) Plasmids: Combining the ease of yeast recombination cloning with the stability of genomic integration. Yeast. 36: 593–605. ArticlePubMedLink
- Fleming E, Yuan AH, Heller DM, Hochschild A. 2019. A bacteria-based genetic assay detects prion formation. Proc Natl Acad Sci USA. 116: 4605–4610. ArticlePubMedPMC
- Frederick KK, Debelouchina GT, Kayatekin C, Dominy T, Jacvone AC, et al. 2014. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem Biol. 21: 295–305. ArticlePubMedPMC
- Gautschi M, Lilie H, Fünfschilling U, Mun A, Ross S, et al. 2001. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc Natl Acad Sci USA. 98: 3762–3767. ArticlePubMedPMC
- Gautschi M, Mun A, Ross S, Rospert S. 2002. A functional chaperone triad on the yeast ribosome. Proc Natl Acad Sci USA. 99: 4209–4214. ArticlePubMedPMC
- Gorkovskiy A, Reidy M, Masison DC, Wickner RB. 2017. Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p. Proc Natl Acad Sci USA. 114: E4193–E4202. ArticlePubMedPMC
- Huang YW, King CY. 2020. A complete catalog of wild-type Sup35 prion variants and their protein-only propagation. Curr Genet. 66: 97–122. ArticlePubMedPDF
- Huang YW, Kushnirov VV, King CY. 2021. Mutable yeast prion variants are stabilized by a defective Hsp104 chaperone. Mol Microbiol. 115: 774–788. ArticlePubMedLink
- Huang VJ, Stein KC, True HL. 2013. Spontaneous variants of the [RNQ+] prion in yeast demonstrate the extensive conformational diversity possible with prion proteins. PLoS One. 8: e79582. ArticlePubMedPMC
- Inge-Vechtomov SG, Tikhodeev ON, Karpova TS. 1988. Selective systems for obtaining recessive ribosomal suppressors in Saccharomyces cerevisiae. Genetika. 24: 1159–1165. PubMed
- Jung G, Jones G, Masison DC. 2002. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 99: 9936–9941. ArticlePubMedPMC
- Jung G, Jones G, Wegrzyn RD, Masison DC. 2000. A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics. 156: 559–570. ArticlePubMedPMCPDF
- Jung G, Masison DC. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol. 43: 7–10. ArticlePubMedPDF
- Kiktev DA, Melomed MM, Lu CD, Newnam GP, Chernoff YO. 2015. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol Microbiol. 96: 621–632. ArticlePubMedPMC
- King CY. 2001. Supporting the structural basis of prion strains: induction and identification of [PSI] variants. J Mol Biol. 307: 1247–1260. ArticlePubMed
- King CY. 2025. Total propagation of yeast prion conformers in ssz1∆ upf1∆ Hsp104T160M triple mutants. Curr Genet. 71: 8.ArticlePubMedPMCPDF
- Kryndushkin D, Shewmaker F, Wickner RB. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 27: 2725–2735. ArticlePubMedPMC
- Lancaster DL, Dobson CM, Rachubinski RA. 2013. Chaperone proteins select and maintain [PIN+] prion conformations in Saccharomyces cerevisiae. J Biol Chem. 288: 1266–1276. ArticlePubMed
- Liebman SW, Stewart JW, Sherman F. 1975. Serine substitutions caused by an ochre suppressor in yeast. J Mol Biol. 94: 595–610. ArticlePubMed
- Manogaran AL, Kirkland KT, Liebman SW. 2006. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae. Yeast. 23: 141–147. ArticlePubMed
- Mathur V, Hong JY, Liebman SW. 2009. Ssa1 overexpression and [PIN+] variants cure [PSI+] by dilution of aggregates. J Mol Biol. 390: 155–167. ArticlePubMedPMC
- Mathur V, Taneja V, Sun Y, Liebman SW. 2010. Analyzing the birth and propagation of two distinct prions, [PSI+] and [Het-s]y, in yeast. Mol Biol Cell. 21: 1449–1461. ArticlePubMedPMC
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. 2005. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA. 102: 10575–10580. ArticlePubMedPMC
- Namy O, Duchateau-Nguyen G, Hatin I, Hermann-Le Denmat S, Termier M, et al. 2003. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 31: 2289–2296. ArticlePubMedPMC
- Ohhashi Y, Yamaguchi Y, Kurahashi H, Kamatari YO, Sugiyama S, et al. 2018. Molecular basis for diversification of yeast prion strain conformation. Proc Natl Acad Sci USA. 115: 2389–2394. ArticlePubMedPMC
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. 1997. Interaction between yeast Sup45p (eRF1) and Sup35p (eRF3) polypeptide chain release factors: implications for prion-dependent regulation. Mol Biol Cell. 17: 2798–2805. ArticleLink
- Pfund C, Lopez-Hoyo N, Ziegelhoffer T, Schilke BA, Lopez-Buesa P, et al. 1998. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 17: 3981–3989. ArticlePubMedPMC
- Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science. 216: 136–144. ArticlePubMed
- Rakwalska M, Rospert S. 2004. The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol Cell Biol. 24: 9186–9197. ArticlePubMedPMCLink
- Roman H. 1956. Studies of gene mutation in Saccharomyces. Cold Spring Harb Symp Quant Biol. 21: 175–185. ArticlePubMed
- Schlumpberger M, Prusiner SB, Herskowitz I. 2001. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 21: 7035–7046. ArticlePubMedPMCLink
- Sharma S, Srivastava S, Dubey RN, Mishra P, Singh J. 2024. [SNG2], a prion form of Cut4/Apc1, confers non-Mendelian inheritance of heterochromatin silencing defect in fission yeast. Nucleic Acids Res. 52: 13792–13811. ArticlePubMedPMCPDF
- Sherman F. 1991. Getting started with yeast. In Guthrie C, Fink GR. (eds.), Guide to yeast genetics and molecular biology, vol. 194, pp. 3–21. Academic Press.Article
- Shewmaker F, Wickner RB, Tycko R. 2006. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc Natl Acad Sci USA. 103: 19754–19759. ArticlePubMedPMC
- Son M. 2024. A story between s and S: [Het-s] prion of the fungus Podospora anserina. Mycobiology. 52: 85–91. ArticlePubMedPMC
- Son M. 2025. Yeast prions: discovery, nature, cellular manipulation and implication. J Microbiol Biotechnol. 35: e2503046. ArticlePubMedPMC
- Son M, Han S, Lee S. 2023. Prions in microbes: the least in the most. J Microbiol. 61: 881–889. ArticlePubMedPDF
- Son M, Wickner RB. 2018. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc Natl Acad Sci USA. 115: E1184–E1193. ArticlePubMedPMC
- Son M, Wickner RB. 2020. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] prion variants. Proc Natl Acad Sci USA. 117: 26298–26306. ArticlePubMedPMC
- Son M, Wickner RB. 2022. Antiprion systems in yeast cooperate to cure or prevent the generation of nearly all [PSI+] and [URE3] prions. Proc Natl Acad Sci USA. 119: e2205500119. ArticlePubMedPMC
- Sondheimer N, Lindquist S. 2000. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 5: 163–172. ArticlePubMed
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, et al. 1995. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 14: 4365–4373. ArticlePubMedPMCLink
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature. 428: 323–328. ArticlePubMedPDF
- Toyama BH, Kelly MJ, Gross JD, Weissman JS. 2007. The structural basis of yeast prion strain variants. Nature. 449: 233–237. ArticlePubMedPDF
- Tuite MF, Mundy CR, Cox BS. 1981. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics. 98: 691–711. ArticlePubMedPMC
- Tycko R. 2014. Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23: 1528–1539. ArticlePubMedPMCLink
- Wickner RB. 1994. [URE3] as an altered URE2 protein: evidence for a prion analog in S. cerevisiae. Science. 264: 566–569. ArticlePubMed
- Wickner RB, Beszonov E, Bateman DA. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci USA. 111: E2711–E2720. ArticlePubMedPMC
- Wickner RB, Dyda F, Tycko R. 2008. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc Natl Acad Sci USA. 105: 2403–2408. ArticlePubMedPMC
- Wickner RB, Kelly AC, Bezsonov EE, Edskes HE. 2017. Prion propagation is controlled by inositol polyphosphates. Proc Natl Acad Sci USA. 114: E8402–E8410. ArticlePubMedPMC
- Wickner RB, Shewmaker F, Bateman DA, Edskes HE, Gorkovskiy A, et al. 2015. Yeast prions: structure, biology and prion-handling systems. Microbiol Mol Biol Rev. 79: 1–17. ArticlePubMedPMCLink
- Wickner RB, Son M, Edskes BK. 2019. Prion variants of yeast are numerous, mutable, and segregate on growth, affecting prion pathogenesis, transmission barriers and sensitivity to anti-prion systems. Viruses. 11: 238.ArticlePubMedPMC
- Wu S, Edskes HK, Wickner RB. 2023. Human proteins curing yeast prions. Proc Natl Acad Sci USA. 120: e2314781120. ArticlePubMedPMC
- Yan W, Schilke B, Pfund C, Walter W, Kim S, et al. 1998. Zuotin, a ribosome‐associated DnaJ molecular chaperone. EMBO J. 17: 4809–4817. ArticlePubMedPMC
- Yu CI, King CY. 2019. Forms and abundance of chaperone proteins influence yeast prion variant competition. Mol Microbiol. 111: 798–810. ArticlePubMedLink
- Yuan AH, Hochschild A. 2017. A bacterial global regulator forms a prion. Science. 355: 198–201. ArticlePubMedPMC
- Zhou P, Derkatch IL, Liebman SW. 2001. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol Microbiol. 39: 37–46. ArticlePubMed
Citations
Citations to this article as recorded by



 ePub Link
ePub Link Cite this Article
Cite this Article