- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 63(10); 2025 > Review
-
Review
Structural analysis of dual specificity phosphatases, the only type of protein tyrosine phosphatases found in humans and across diverse microorganisms - Bonsu Ku*
-
Journal of Microbiology 2025;63(10):e2506006.
DOI: https://doi.org/10.71150/jm.2506006
Published online: October 31, 2025
Orphan Disease Therapeutic Target Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 34141, Republic of Korea
- *Correspondence Bonsu Ku bku@kribb.re.kr
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Dual specificity phosphatases (DUSPs), a subfamily of the protein tyrosine phosphatase (PTP) family, dephosphorylate not only phosphotyrosine but also phosphoserine and phosphothreonine residues. Beyond the 26 members of this family in humans, DUSPs represent the only type of PTPs found across a wide range of microorganisms, including bacteria, archaea, and viruses. This review presents a comprehensive structural analysis of human and microbial DUSPs. These proteins commonly share core features, such as a typical DUSP fold, shallow active site pocket, signature active site motif known as the P-loop, and conserved aspartate residue that acts as a general acid/base. However, DUSPs from diverse microorganisms also display unique structural and functional characteristics. Pseudomonas aeruginosa TpbA is the only bacterial DUSP identified to date, while a second candidate was proposed in this review. Archaeal DUSPs are hyperthermostable, contain a unique motif in their P-loops, and employ dual general acid/base residues. Poxviral DUSPs are characterized by the formation of domain-swapped homodimers. The presence of DUSPs across all domains of life and viruses, along with their low specificity for phosphorylated amino acids and structural similarity to classical PTPs, suggests that DUSPs represent the ancestral form of PTPs.
Introduction
Structural Analysis of the Catalytic Domain of Human DUSPs
Structural Analysis of Microbial DUSPs
Conclusion
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (RS-2023-00278696) and Korea Research Institute of Bioscience and Biotechnology Research Initiative Programs (KGM9952522 and KGM1322511).
Conflict of Interest
The author declares no competing interests exist.
Supplementary Information
| Protein | Alias | PDB code | References |
|---|---|---|---|
| DUSP1 | MKP-1, VH1 | 6APX, 6D65, 6D66, 6D67 | Gumpena et al. (2018a, 2018b) |
| DUSP2 | PAC-1 | 1M3G* | Farooq et al. (2003) |
| DUSP4 | MKP-2, VH2 | 3EZZ | Jeong et al. (2009) |
| DUSP5 | VH3 | 2G6Z | Jeong et al. (2007) |
| DUSP6 | MKP-3, Pyst1 | 1MKP | Farooq et al. (2001); Stewart et al. (1999) |
| DUSP7 | MKP-X, Pyst2 | 4Y2E | Lountos et al. (2015a) |
| DUSP8 | VH5 | 4JMK | Jeong et al. (2014) |
| DUSP9 | MKP-4, Pyst3 | 2HXP, 3LJ8 | Almo et al. (2007); Jeong et al. (2011) |
| DUSP10 | MKP-5 | 1ZZW, 2OUD, 6MC1, 7U4O, 7U4R, 7UMU, 7UMV, 7UN0, 7UN4, 7Y4B, 7Y4C, 7Y4D, 7Y4E | Gannam et al. (2020, 2022); Jeong et al. (2006b); Tao and Tong (2007); Zhang et al. (2011) |
| DUSP14 | MKP-6 | 2WGP | Lountos et al. (2009) |
| DUSP16 | MKP-7 | 4YR8 | Liu et al. (2016); Zhang et al. (2011) |
| Protein | Alias | PDB code | References |
|---|---|---|---|
| DUSP3 | VHR | 1J4X, 1VHR, 3F81, 8TK2, 8TK3, 8TK4, 8TK5, 8TK6, 9DJ9 | Schumacher et al. (2002); Wu et al. (2009, 2025); Yuvaniyama et al. (1996) |
| DUSP11 | PIR1 | 4JMJ, 4MBB, 4NYH | Jeong et al. (2014); Sankhala et al. (2014) |
| DUSP12 | YVH1 | 4JNB, 4KI9 | Jeong et al. (2014) |
| DUSP13a | BEDP | 5XJV | Wei et al. (2018) |
| DUSP13b | SKRP4, TMDP | 2GWO, 2PQ5 | Kim et al. (2007) |
| DUSP15 | VHY | 1YZ4 | Yoon et al. (2005) |
| DUSP18 | DUSP20 | 2ESB | Jeong et al. (2006a) |
| DUSP19 | DUSP17, SKRP1 | 3S4E, 4D3P, 4D3Q, 4D3R | Jeon et al. (2015); Wei et al. (2011) |
| DUSP21 | - | (none) | (none) |
| DUSP22 | JSP1, VHX | 1WRM, 4WOH, 6L1S, 6LMY, 6LOT, 6LOU, 6LVQ, 7C8S | Lai et al. (2020); Lountos et al. (2015b); Yokota et al. (2007) |
| DUSP23a | VHZ, DUSP25 | 2IMG, 4ERC | Agarwal et al. (2008); Kuznetsov et al. (2012) |
| DUSP23b | PTPMT1, MOSP | (none) | (none) |
| DUSP26 | DUSP24, SKRP3 | 2E0T, 4B04, 4HRF, 5GTJ | Lokareddy et al. (2013); Won et al. (2013, 2016) |
| DUSP28 | VHP | 5Y15, 5Y16 | Ku et al. (2017) |
| DUSP29 | DUSP27. DUPD1 | 2Y96 | Lountos et al. (2011) |
| Classification | Species | Protein | PDB code | References |
|---|---|---|---|---|
| Bacteria | P. aeruginosa | TpbA | 2M3V*, 4R0S, 4R0T | Koveal et al. (2013); Xu et al. (2015) |
| Archaea | T. kodakarensis | Tk-PTP | 5Z59, 5Z5A, 5Z5B | Yun et al. (2018) |
| S. solfataricus | SsoPTP | 2DXP, 2I6I, 2I6J, 2I6M, 2I6O, 2I6P, 7MPC, PMPD, | Chu and Wang (2007); Pinkston et al. (2021) | |
| Virus | Variola virus | H1 | 2P4D | Phan et al. (2007) |
| Vaccinia virus | VH1 | 2Q05, 2RF6, 3CM3 | Koksal et al. (2009) | |
| Monkeypox virus | H1 | 8GZ4 | Cui et al. (2023) | |
| Orf virus | OH1 | 5NCR | Segovia et al. (2017) |
- Agarwal R, Burley SK, Swaminathan S. 2008. Structure of human dual specificity protein phosphatase 23 VHZ enzyme-substrate/product complex. J Biol Chem. 283: 8946–8953. ArticlePubMed
- Almo SC, Bonanno JB, Sauder JM, Emtage S, Dilorenzo TP, et al. 2007. Structural genomics of protein phosphatases. J Struct Funct Genomics. 8: 121–140. ArticlePubMedPMCPDF
- Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. 2017. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med. 40: 271–280. ArticlePubMedPMC
- Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. 2008. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human Vacuolar Protein Sorting 33B. Cell Host Microbe. 3: 316–322. ArticlePubMed
- Braicu C, Buse M, Busuioc C, Drula R, Gulei D, et al. 2019. A comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers. 11: 1618.ArticlePubMedPMC
- Caunt CJ, Keyse SM. 2013. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 280: 489–504. ArticlePubMedPMC
- Chu HM, Wang AHJ. 2007. Enzyme-substrate interactions revealed by the crystal structures of the archaeal Sulfolobus PTP-fold phosphatase and its phosphopeptide complexes. Proteins. 66: 996–1003. ArticlePubMed
- Cui W, Huang H, Duan Y, Luo Z, Wang H, et al. 2023. Crystal structure of monkeypox H1 phosphatase, an antiviral drug target. Protein Cell. 14: 469–472. ArticlePubMedPDF
- Diggle SP, Whiteley M. 2020. Microbe profile: Pseudomonas aeruginosa: opportunistic pathogen and lab rat. Microbiology. 166: 30–33. ArticlePubMed
- Esser D, Hoffmann L, Pham TK, Bräsen C, Qiu W, et al. 2016. Protein phosphorylation and its role in archaeal signal transduction. FEMS Microbiol Rev. 40: 625–647. ArticlePubMedPMC
- Farooq A, Chaturvedi G, Mujtaba S, Plotnikova O, Zeng L, et al. 2001. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3. Mol Cell. 7: 387–399. ArticlePubMed
- Farooq A, Plotnikova O, Chaturvedi G, Yan S, Zeng L, et al. 2003. Solution structure of the MAPK phosphatase PAC-1 catalytic domain. Structure. 11: 155–164. ArticlePubMed
- Fjeld CC, Rice AE, Kim Y, Gee KR, Denu JM. 2000. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J Biol Chem. 275: 6749–6757. ArticlePubMed
- Gannam ZTK, Jamali H, Kweon OS, Herrington J, Shillingford SR, et al. 2022. Defining the structure-activity relationship for a novel class of allosteric MKP5 inhibitors. Eur J Med Chem. 243: 114712.ArticlePubMedPMC
- Gannam ZTK, Min K, Shillingford SR, Zhang L, Herrington J, et al. 2020. An allosteric site on MKP5 reveals a strategy for small-molecule inhibition. Sci Signal. 13: eaba3043. ArticlePubMedPMC
- Guan KL, Dixon JE. 1993. Bacterial and viral protein tyrosine phosphatases. Semin Cell Biol. 4: 389–396. ArticlePubMed
- Gumpena R, Lountos GT, Raran-Kurussi S, Tropea JE, Cherry S, et al. 2018a. Crystal structure of the human dual specificity phosphatase 1 catalytic domain. Protein Sci. 27: 561–567. ArticleLink
- Gumpena R, Lountos GT, Waugh DS. 2018b. MBP-binding DARPins facilitate the crystallization of an MBP fusion protein. Acta Crystallogr F Struct Biol Commun. 74: 549–557. Article
- Hong SB, Lubben TH, Dolliver CM, Petrolonis AJ, Roy RA, et al. 2005. Expression, purification, and enzymatic characterization of the dual specificity mitogen-activated protein kinase phosphatase MKP-4. Bioorg Chem. 33: 34–44. ArticlePubMed
- Humphreys D, Hume PJ, Koronakis V. 2009. The Salmonella effector SptP dephosphorylates host AAA+ ATPase VCP to promote development of its intracellular replicative niche. Cell Host Microbe. 5: 225–233. ArticlePubMedPMC
- Jeon TJ, Nam KT, Ryu SE. 2015. Structural analysis of activity-modulating mutations of DUSP19. Biodesign. 3: 111–116.
- Jeong DG, Cho YH, Yoon TS, Kim JH, Ryu SE, et al. 2007. Crystal structure of the catalytic domain of human DUSP5 a dual specificity MAP kinase protein phosphatase. Proteins. 66: 253–258. ArticlePubMed
- Jeong DG, Cho YH, Yoon TS, Kim JH, Son JH, et al. 2006a. Structure of human DSP18 a member of the dual-specificity protein tyrosine phosphatase family. Acta Crystallogr D Biol Crystallogr. 62: 582–588. Article
- Jeong DG, Jung SK, Yoon TS, Woo EJ, Kim JH, et al. 2009. Crystal structure of the catalytic domain of human MKP-2 reveals a 24-mer assembly. Proteins. 76: 763–767. ArticlePubMed
- Jeong DG, Wei CH, Ku B, Jeon TJ, Chien PN, et al. 2014. The family-wide structure and function of human dual-specificity protein phosphatases. Acta Crystallogr D Biol Crystallogr. 70: 421–435. ArticlePubMed
- Jeong DG, Yoon TS, Jung SK, Park BC, Park H, et al. 2011. Exploring binding sites other than the catalytic core in the crystal structure of the catalytic domain of MKP-4. Acta Crystallogr D Biol Crystallogr. 67: 25–31. ArticlePubMed
- Jeong DG, Yoon TS, Kim JH, Shim MY, Jung SK, et al. 2006b. Crystal structure of the catalytic domain of human MAP kinase phosphatase 5: structural insight into constitutively active phosphatase. J Mol Biol. 360: 946–955. Article
- Kant S, Agarwal S, Pancholi P, Pancholi V. 2015. The Streptococcus pyogenes orphan protein tyrosine phosphatase SP-PTP possesses dual specificity and essential virulence regulatory functions. Mol Microbiol. 97: 515–540. ArticlePubMed
- Kant S, Pancholi V. 2021. Novel tyrosine kinase-mediated phosphorylation with dual specificity plays a key role in the modulation of Streptococcus pyogenes physiology and virulence. Front Microbiol. 12: 689246.ArticlePubMedPMC
- Kennelly PJ. 2014. Protein Ser/Thr/Tyr phosphorylation in the Archaea. J Biol Chem. 289: 9480–9487. ArticlePubMedPMC
- Kim SJ, Jeong DG, Yoon TS, Son JH, Cho SK, et al. 2007. Crystal structure of human TMDP a testis-specific dual specificity protein phosphatase: implications for substrate specificity. Proteins. 66: 239–245. ArticlePubMed
- Koksal AC, Cingolani G. 2011. Dimerization of vaccinia virus VH1 is essential for dephosphorylation of STAT1 at tyrosine 701. J Biol Chem. 286: 14373–14382. ArticlePubMedPMC
- Koksal AC, Nardozzi JD, Cingolani G. 2009. Dimeric quaternary structure of the prototypical dual specificity phosphatase VH1. J Biol Chem. 284: 10129–10137. ArticlePubMedPMC
- Kolmodin K, Aqvist J. 2001. The catalytic mechanism of protein tyrosine phosphatases revisited. FEBS Lett. 498: 208–213. ArticlePubMedLink
- Kondoh K, Nishida E. 2007. Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta. 1773: 1227–1237. ArticlePubMed
- Koveal D, Clarkson MW, Wood TK, Page R, Peti W. 2013. Ligand binding reduces conformational flexibility in the active site of tyrosine phosphatase related to biofilm formation A (TpbA) from Pseudomonas aeruginosa. J Mol Biol. 425: 2219–2231. ArticlePubMedPMC
- Ku B, Hong W, Keum CW, Kim M, Ryu H, et al. 2017. Structural and biochemical analysis of atypically low dephosphorylating activity of human dual-specificity phosphatase 28. PLoS One. 12: e0187701. ArticlePubMedPMC
- Ku B, Keum CW, Lee HS, Yun HY, Shin HC, et al. 2016. Crystal structure of SP-PTP a low molecular weight protein tyrosine phosphatase from Streptococcus pyogenes. Biochem Biophys Res Commun. 478: 1217–1222. ArticlePubMed
- Kuban-Jankowska A, Kostrzewa T, Gorska-Ponikowska M. 2022. Bacterial protein tyrosine phosphatases as possible targets for antimicrobial therapies in response to antibiotic resistance. Antioxidants. 11: 2397.ArticlePubMedPMC
- Kuznetsov VI, Hengge AC. 2013. New functional aspects of the atypical protein tyrosine phosphatase VHZ. Biochemistry. 52: 8012–8025. ArticlePubMed
- Kuznetsov VI, Hengge AC, Johnson SJ. 2012. New aspects of the phosphatase VHZ revealed by a high-resolution structure with vanadate and substrate screening. Biochemistry. 51: 9869–9879. ArticlePubMed
- Lai CH, Chang CC, Chuang HC, Tan TH, Lyu PC. 2020. Structural insights into the active site formation of DUSP22 in N-loop-containing protein tyrosine phosphatases. Int J Mol Sci. 21: 7515.ArticlePubMedPMC
- Lee H, Yi JS, Lawan A, Min K, Bennett AM. 2015. Mining the function of protein tyrosine phosphatases in health and disease. Semin Cell Dev Biol. 37: 66–72. ArticlePubMed
- Liu X, Zhang CS, Lu C, Lin SC, Wu JW, et al. 2016. A conserved motif in JNK/p38-specific MAPK phosphatases as a determinant for JNK1 recognition and inactivation. Nat Commun. 7: 10879.ArticlePubMedPMCPDF
- Lokareddy RK, Bhardwaj A, Cingolani G. 2013. Atomic structure of dual-specificity phosphatase 26, a novel p53 phosphatase. Biochemistry. 52: 938–948. ArticlePubMed
- Lountos GT, Austin BP, Tropea JE, Waugh DS. 2015a. Structure of human dual-specificity phosphatase 7, a potential cancer drug target. Acta Crystallogr F Struct Biol Commun. 71: 650–656. Article
- Lountos GT, Cherry S, Tropea JE, Waugh DS. 2015b. Structural analysis of human dual-specificity phosphatase 22 complexed with a phosphotyrosine-like substrate. Acta Crystallogr F Struct Biol Commun. 71: 199–205. Article
- Lountos GT, Tropea JE, Cherry S, Waugh DS. 2009. Overproduction purification and structure determination of human dual-specificity phosphatase 14. Acta Crystallogr D Biol Crystallogr. 65: 1013–1020. ArticlePubMedPMC
- Lountos GT, Tropea JE, Waugh DS. 2011. Structure of human dual-specificity phosphatase 27 at 2.38 Å resolution. Acta Crystallogr D Biol Crystallogr. 67: 471–479. ArticlePubMedPMC
- Mann BA, Huang JH, Li P, Chang HC, Slee RB, et al. 2008. Vaccinia virus blocks Stat1-dependent and Stat1-independent gene expression induced by type I and type II interferons. J Interferon Cytokine Res. 28: 367–380. ArticlePubMedPMC
- Morikawa M, Izawa Y, Rashid N, Hoaki T, Imanaka T. 1994. Purification and characterization of a thermostable thiol protease from a newly isolated hyperthermophilic Pyrococcus sp. Appl Environ Microbiol. 60: 4559–4566. ArticlePubMedPMCLink
- Najarro P, Traktman P, Lewis JA. 2001. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol. 75: 3185–3196. ArticlePubMedPMCLink
- Patterson KI, Brummer T, O'Brien PM, Daly RJ. 2009. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 418: 475–489. ArticlePubMedPDF
- Phan J, Tropea JE, Waugh DS. 2007. Structure-assisted discovery of Variola major H1 phosphatase inhibitors. Acta Crystallogr D Biol Crystallogr. 63: 698–704. ArticlePubMed
- Pinkston J, Jo J, Olsen KJ, Comer D, Glaittli CA, et al. 2021. Significant loop motions in the SsoPTP protein tyrosine phosphatase allow for dual general acid functionality. Biochemistry. 60: 2888–2901. ArticlePubMed
- Pu M, Wood TK. 2010. Tyrosine phosphatase TpbA controls rugose colony formation in Pseudomonas aeruginosa by dephosphorylating diguanylate cyclase TpbB. Biochem Biophys Res Commun. 402: 351–355. ArticlePubMedPMC
- Sankhala RS, Lokareddy RK, Cingolani G. 2014. Structure of human PIR1, an atypical dual-specificity phosphatase. Biochemistry. 53: 862–871. ArticlePubMed
- Schumacher MA, Todd JL, Rice AE, Tanner KG, Denu JM. 2002. Structural basis for the recognition of a bisphosphorylated MAP kinase peptide by human VHR protein phosphatase. Biochemistry. 41: 3009–3017. ArticlePubMed
- Segovia D, Haouz A, Porley D, Olivero N, Martinez M, et al. 2017. OH1 from Orf virus: a new tyrosine phosphatase that displays distinct structural features and triple substrate specificity. J Mol Biol. 429: 2816–2824. ArticlePubMed
- Selner NG, Luechapanichkul R, Chen X, Neel BG, Zhang ZY, et al. 2014. Diverse levels of sequence selectivity and catalytic efficiency of protein-tyrosine phosphatases. Biochemistry. 53: 397–412. ArticlePubMed
- Sossai P, Staiti D, Cannas M, Grima P. 2023. Smallpox and monkeypox: looking back and looking ahead. Cleve Clin J Med. 90: 141–144. ArticlePubMed
- Sprygin A, Mazloum A, van Schalkwyk A, Babiuk S. 2022. Capripoxviruses, leporipoxviruses, and orthopoxviruses: occurrences of recombination. Front Microbiol. 13: 978829.ArticlePubMedPMC
- Standish AJ, Morona R. 2014. The role of bacterial protein tyrosine phosphatases in the regulation of the biosynthesis of secreted polysaccharides. Antioxid Redox Signal. 20: 2274–2289. ArticlePubMedPMC
- Stewart AE, Dowd S, Keyse SM, McDonald NQ. 1999. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. Nat Struct Biol. 6: 174–181. ArticlePubMed
- Stravopodis DJ, Kyrpides NC. 1999. Identification of protein-tyrosine phosphatases in Archaea. J Mol Evol. 48: 625–627. ArticlePubMedPDF
- Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. 2008. Protein tyrosine phosphatases: structure-function relationships. FEBS J. 275: 867–882. ArticlePubMed
- Tao X, Tong L. 2007. Crystal structure of the MAP kinase binding domain and the catalytic domain of human MKP5. Protein Sci. 16: 880–886. ArticlePubMedPMC
- Tarrant MK, Cole PA. 2009. The chemical biology of protein phosphorylation. Annu Rev Biochem. 78: 797–825. ArticlePubMedPMC
- Tautz L, Critton DA, Grotegut S. 2013. Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol Biol. 1053: 179–221. ArticlePubMedPMC
- Tonks NK. 2006. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 7: 833–846. ArticlePubMedPDF
- Ubersax JA, Ferrell JE Jr. 2007. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 8: 530–541. ArticlePubMedPDF
- Wang X, Liu M, Yu C, Li J, Zhou X. 2023. Biofilm formation: mechanistic insights and therapeutic targets. Mol Biomed. 4: 49.ArticlePubMedPMCPDF
- Wei CH, Min HG, Kim M, Kim GH, Chun HJ, et al. 2018. Two intermediate states of the conformational switch in dual specificity phosphatase 13a. Pharmacol Res. 128: 211–219. ArticlePubMed
- Wei CH, Ryu SY, Jeon YH, Yoon MY, Jeong DG, et al. 2011. Crystal structure of a novel mitogen-activated protein kinase phosphatase, SKRP1. Proteins. 79: 3242–3246. ArticlePubMedLink
- Wenta N, Strauss H, Meyer S, Vinkemeier U. 2008. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci USA. 105: 9238–9243. ArticlePubMedPMC
- Whitmore SE, Lamont RJ. 2012. Tyrosine phosphorylation and bacterial virulence. Int J Oral Sci. 4: 1–6. ArticlePubMedPMCPDF
- Won EY, Lee SO, Lee DH, Lee D, Bae KH, et al. 2016. Structural insight into the critical role of the N-terminal region in the catalytic activity of dual-specificity phosphatase 26. PLoS One. 11: e0162115. ArticlePubMedPMC
- Won EY, Xie Y, Takemoto C, Chen L, Liu ZJ, et al. 2013. High-resolution crystal structure of the catalytic domain of human dual-specificity phosphatase 26. Acta Crystallogr D Biol Crystallogr. 69: 1160–1170. ArticlePubMed
- Wu J, Baranowski MR, Aleshin AE, Isiorho EA, Lambert LJ, et al. 2025. Fragment screening identifies novel allosteric binders and binding sites in the VHR (DUSP3) phosphatase. ACS Omega. 10: 4912–4926. ArticlePubMedPMCLink
- Wu S, Vossius S, Rahmouni S, Miletic AV, Vang T, et al. 2009. Multidentate small-molecule inhibitors of vaccinia H1-related (VHR) phosphatase decrease proliferation of cervix cancer cells. J Med Chem. 52: 6716–6723. ArticlePubMedPMC
- Xu K, Li S, Yang W, Li K, Bai Y, et al. 2015. Structural and biochemical analysis of tyrosine phosphatase related to biofilm formation A (TpbA) from the opportunistic pathogen Pseudomonas aeruginosa PAO1. PLoS One. 10: e0124330. ArticlePubMedPMC
- Yokota T, Nara Y, Kashima A, Matsubara K, Misawa S, et al. 2007. Crystal structure of human dual specificity phosphatase, JNK stimulatory phosphatase-1, at 1.5 Å resolution. Proteins. 66: 272–278. ArticlePubMed
- Yoon TS, Jeong DG, Kim JH, Cho YH, Son JH, et al. 2005. Crystal structure of the catalytic domain of human VHY, a dual-specificity protein phosphatase. Proteins. 61: 694–697. ArticlePubMed
- Yu H, Bruneau RC, Brennan G, Rothenburg S. 2021. Battle royale: innate recognition of poxviruses and viral immune evasion. Biomedicines. 9: 765.ArticlePubMedPMC
- Yun HY, Kim MW, Lee HS, Kim W, Shin JH, et al. 2019. Structural basis for recognition of the tumor suppressor protein PTPN14 by the oncoprotein E7 of human papillomavirus. PLoS Biol. 17: e3000367. ArticlePubMedPMC
- Yun HY, Lee J, Kim H, Ryu H, Shin HC, et al. 2018. Structural study reveals the temperature-dependent conformational flexibility of Tk-PTP, a protein tyrosine phosphatase from Thermococcus kodakaraensis KOD1. PLoS One. 13: e0197635. ArticlePubMedPMC
- Yuvaniyama J, Denu JM, Dixon JE, Saper MA. 1996. Crystal structure of the dual specificity protein phosphatase VHR. Science. 272: 1328–1331. ArticlePubMed
- Zhang ZY. 2002. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 42: 209–234. ArticlePubMed
- Zhang ZY, Wang Y, Dixon JE. 1994. Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc Natl Acad Sci USA. 91: 1624–1627. ArticlePubMedPMC
- Zhang YY, Wu JW, Wang ZX. 2011. A distinct interaction mode revealed by the crystal structure of the kinase p38α with the MAPK binding domain of the phosphatase MKP5. Sci Signal. 4: ra88.ArticlePubMed
- Zhou B, Zhang ZY. 1999. Mechanism of mitogen-activated protein kinase phosphatase-3 activation by ERK2. J Biol Chem. 274: 35526–35534. ArticlePubMed
References
Supplementary Information
References
Citations
- DUSP family phosphatases in cell signaling, inflammation, and chronic diseases
Chia-Wen Wang, Huai-Chia Chuang, Tse-Hua Tan
Journal of Biomedical Science.2026;[Epub] CrossRef - Structural and biochemical analyses of a novel bacterial dual specificity phosphatase from Candidatus Chlorohelix allophototropha
Sujin Jung, So Hyeon Park, Joon Sig Choi, Ho-Chul Shin, Seung Jun Kim, Bonsu Ku
Journal of Microbiology.2026; 64(7): e2604025. CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article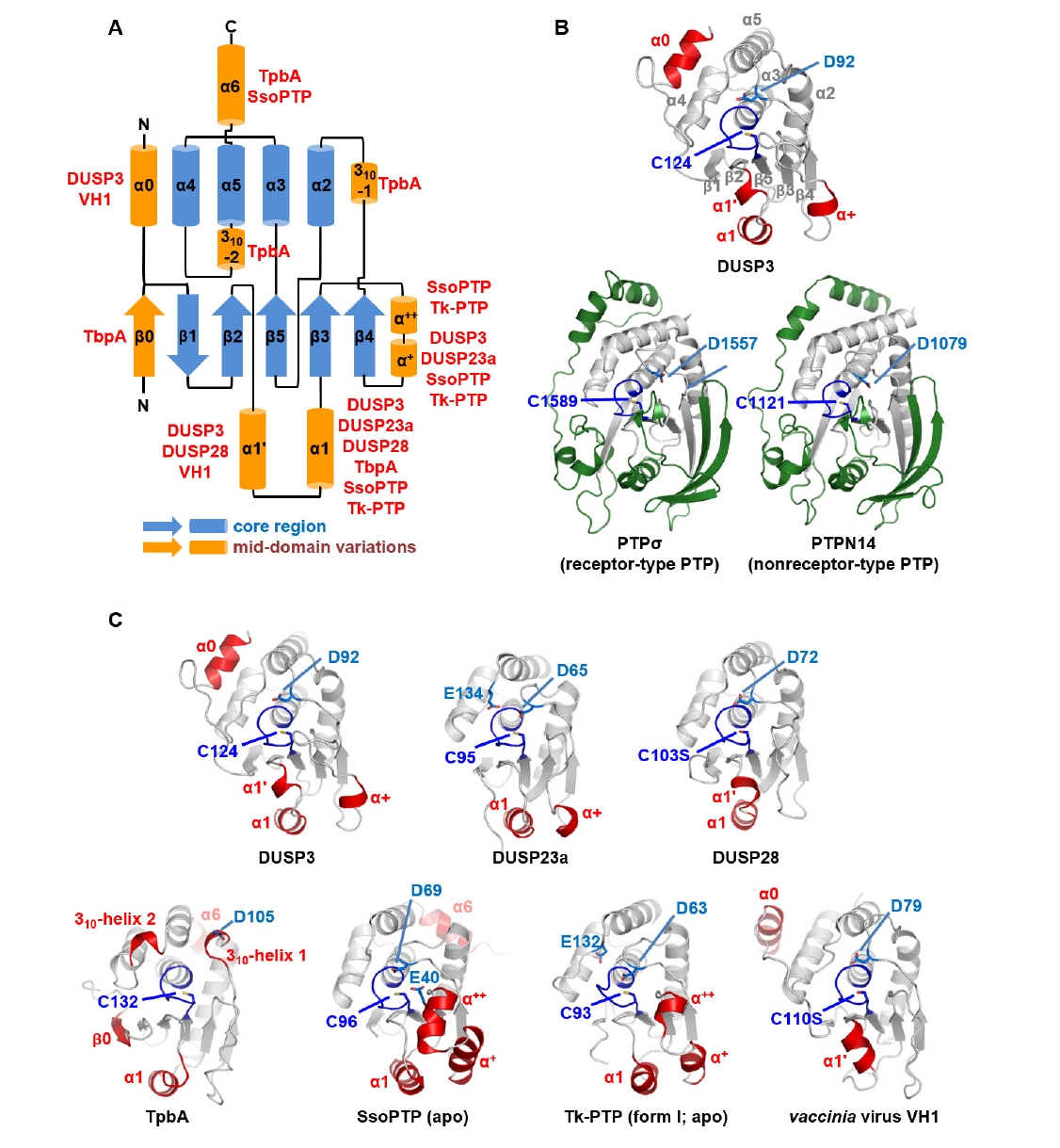
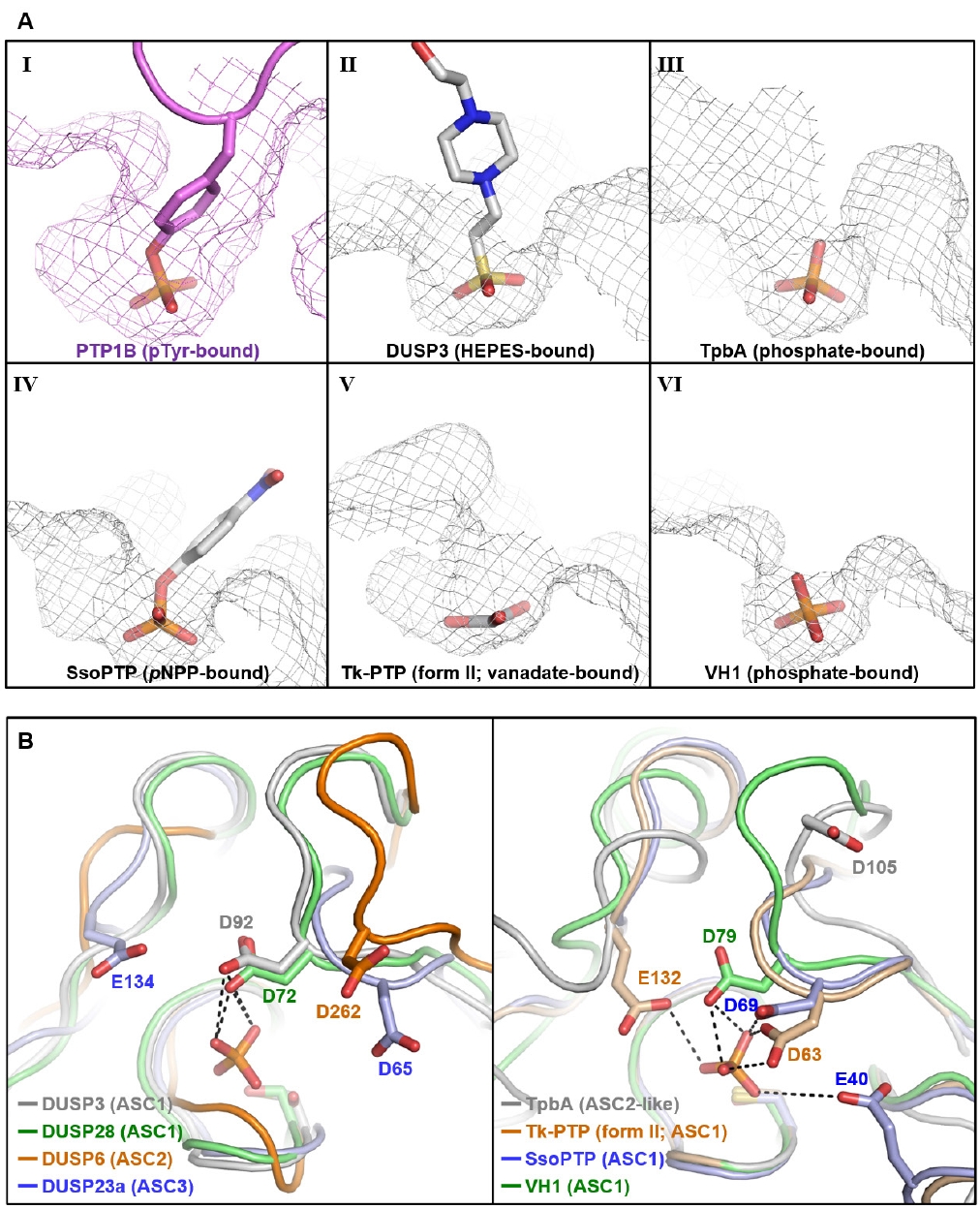
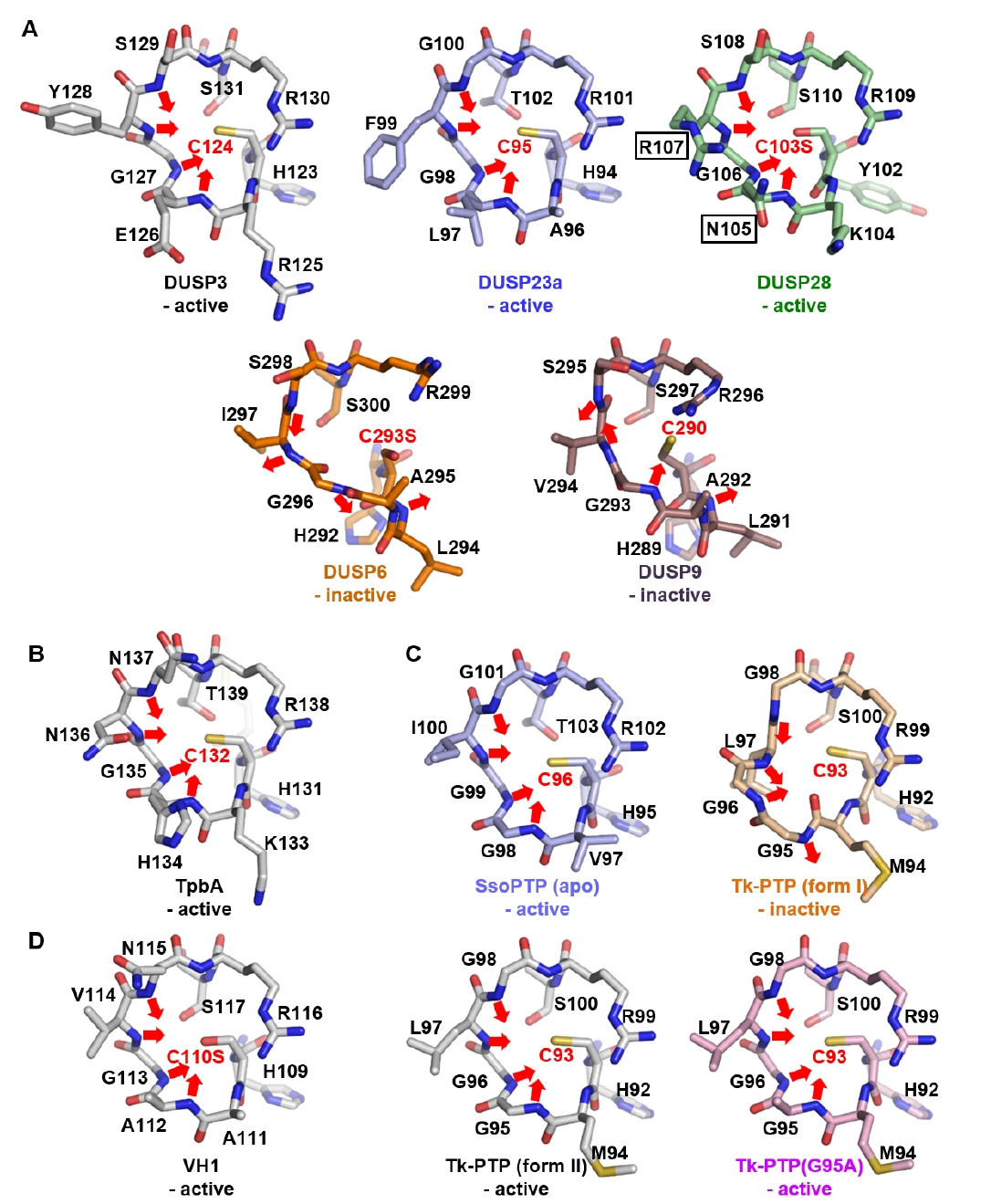
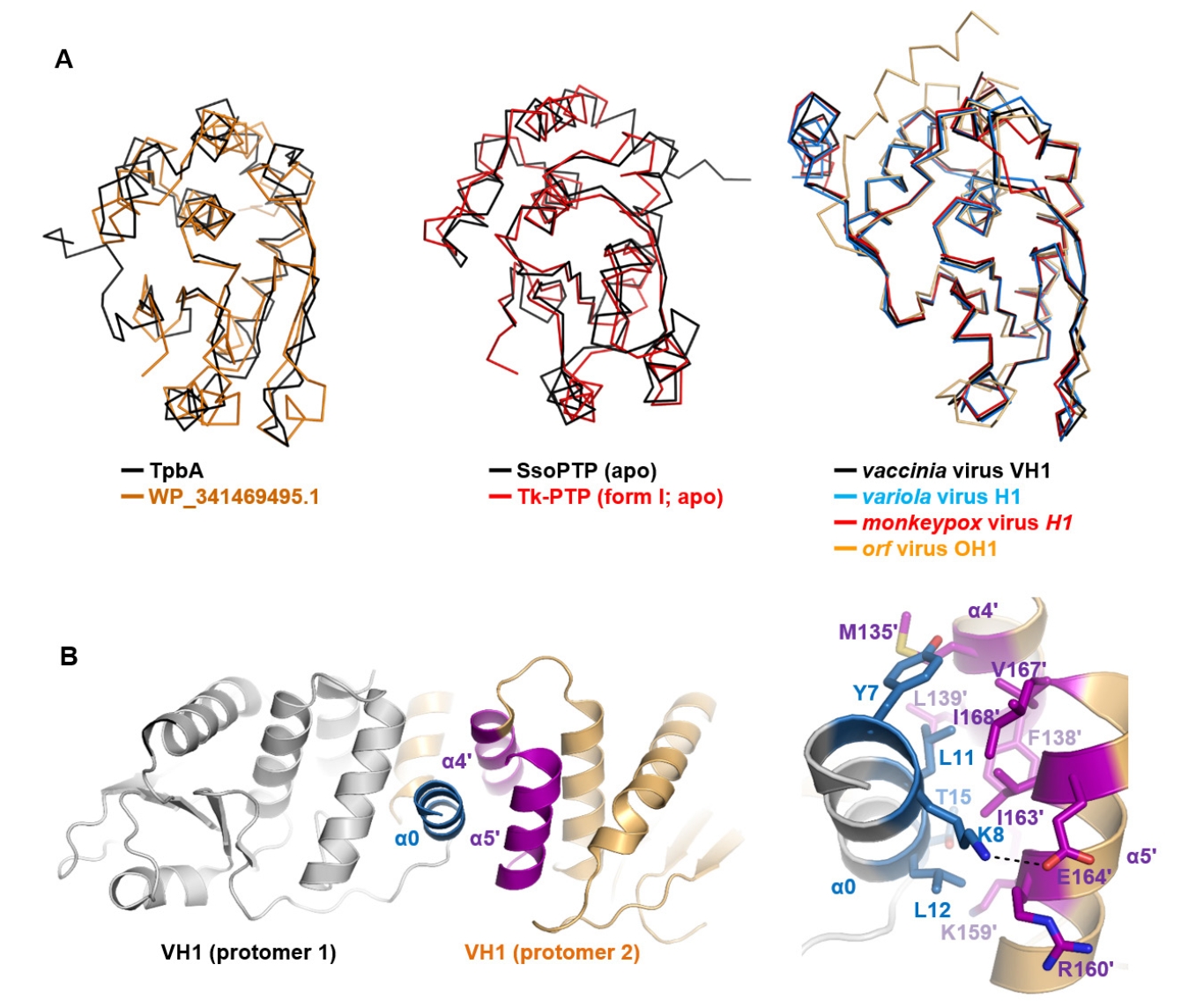




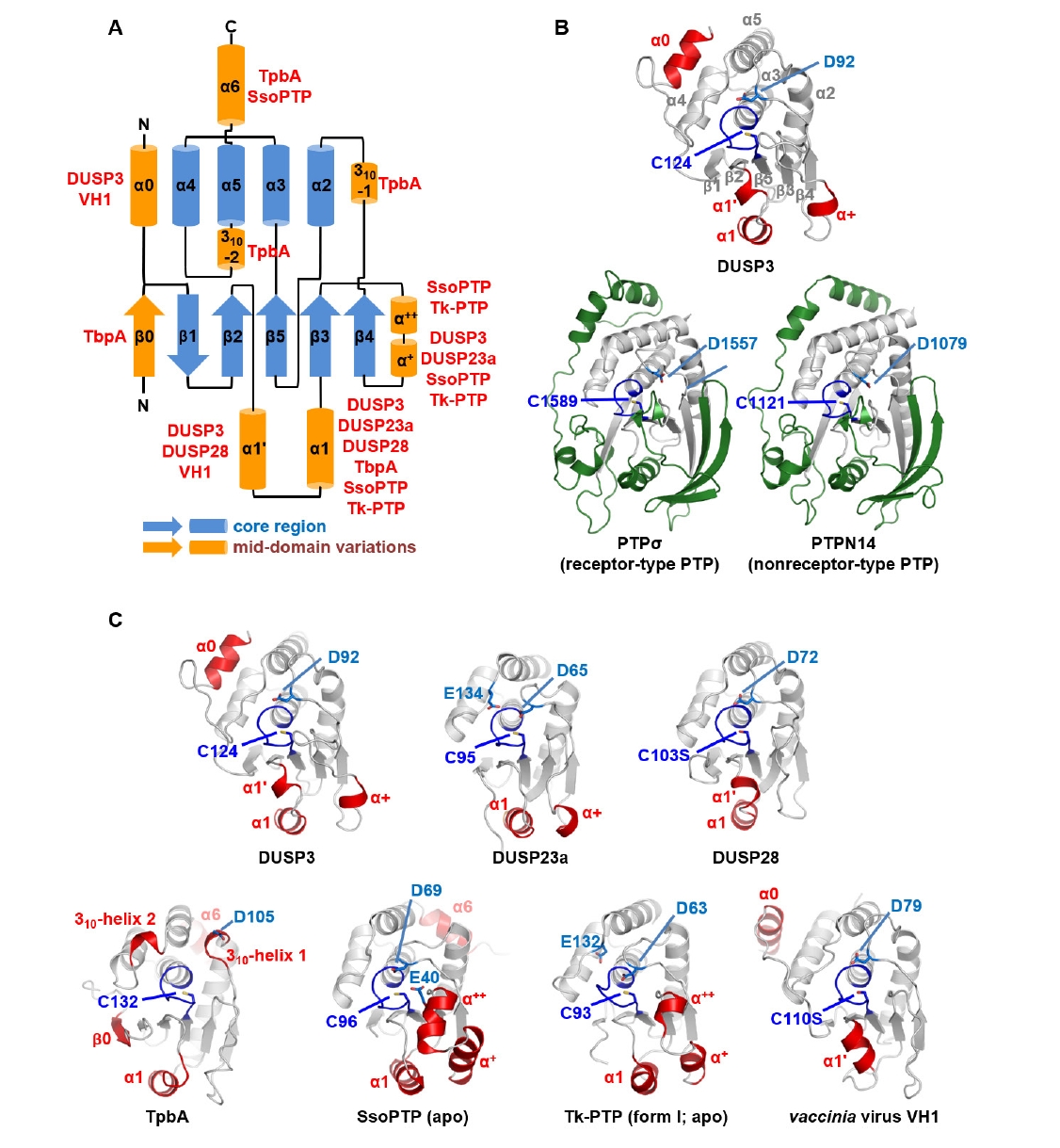
Fig. 1.
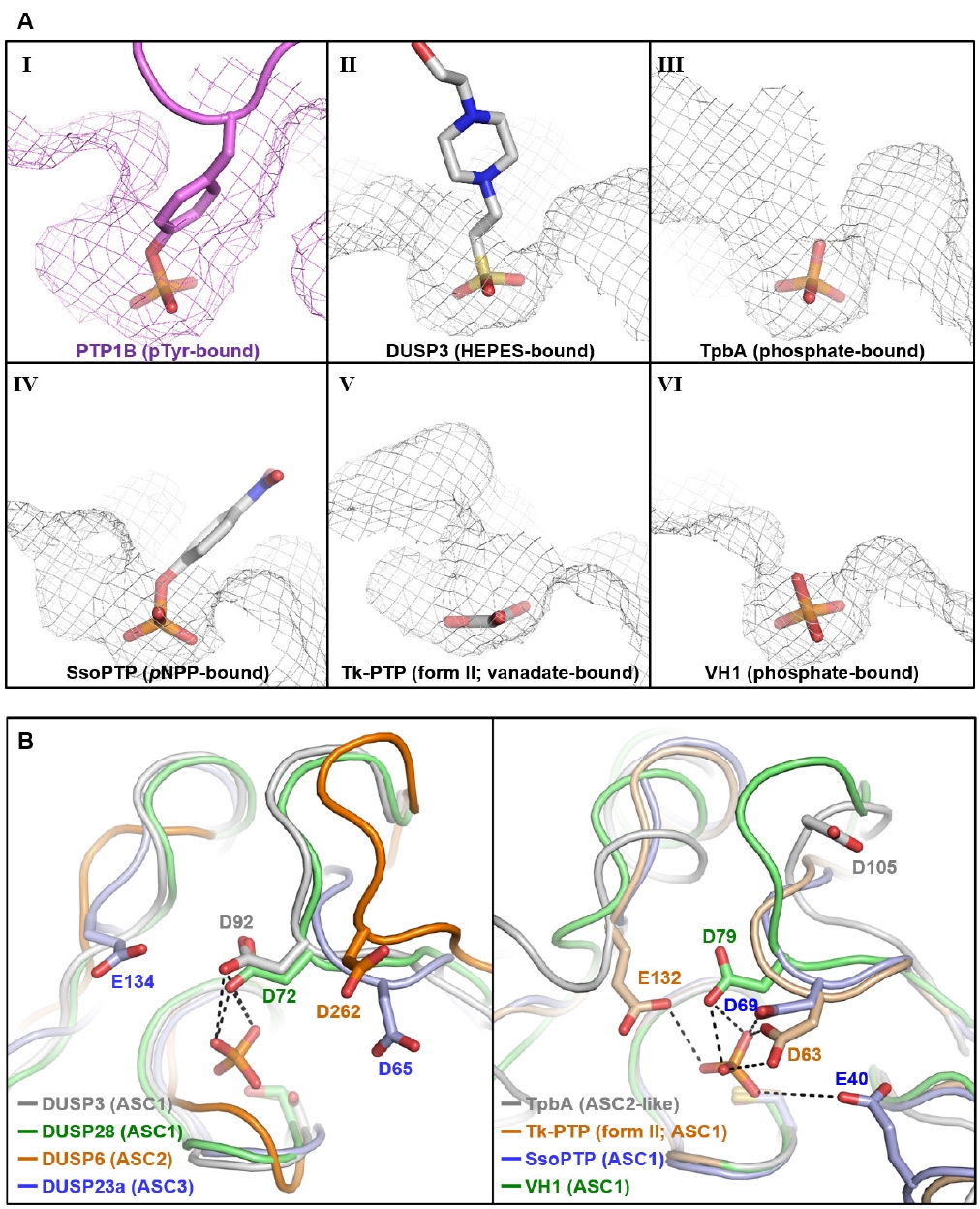
Fig. 2.
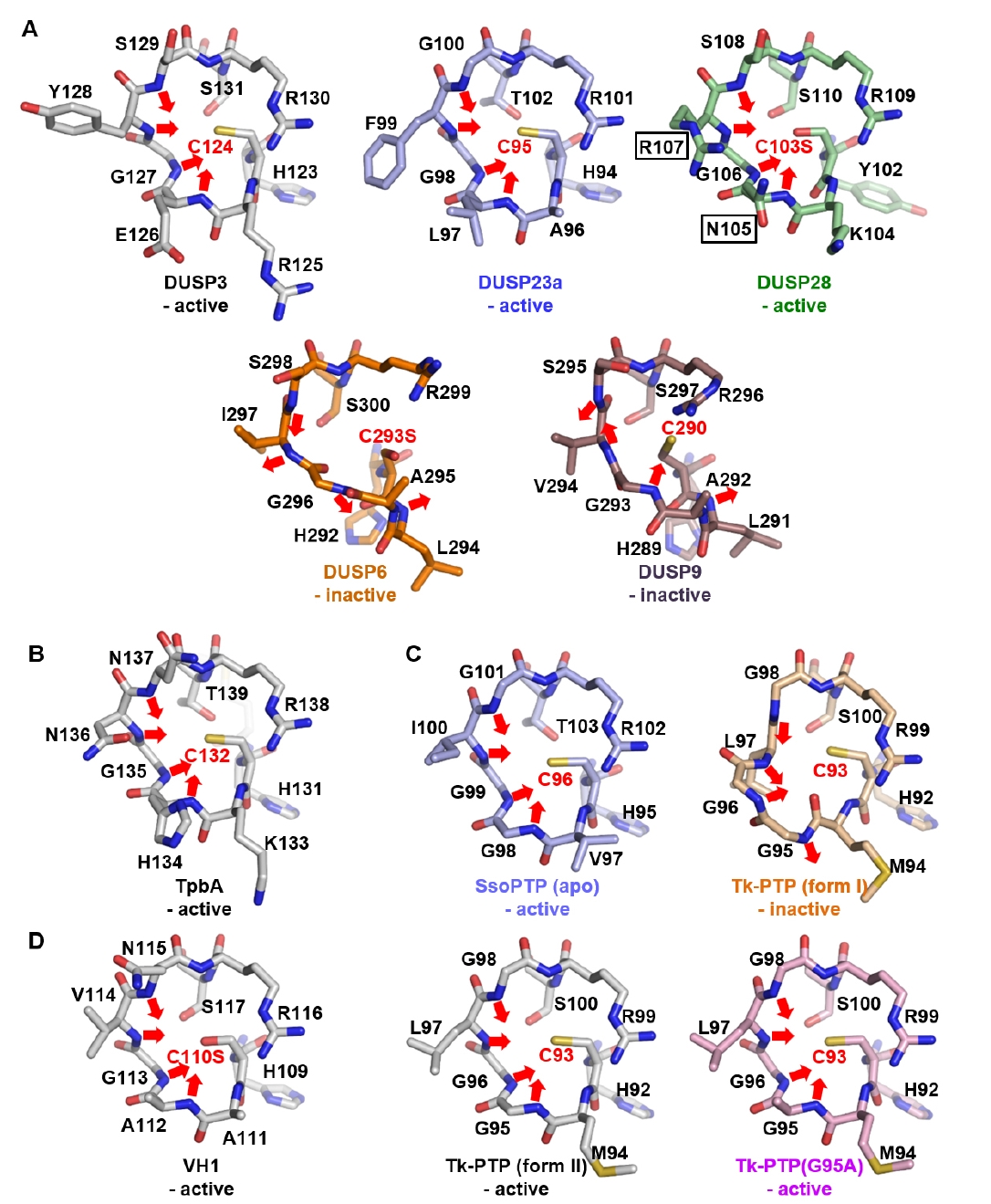
Fig. 3.
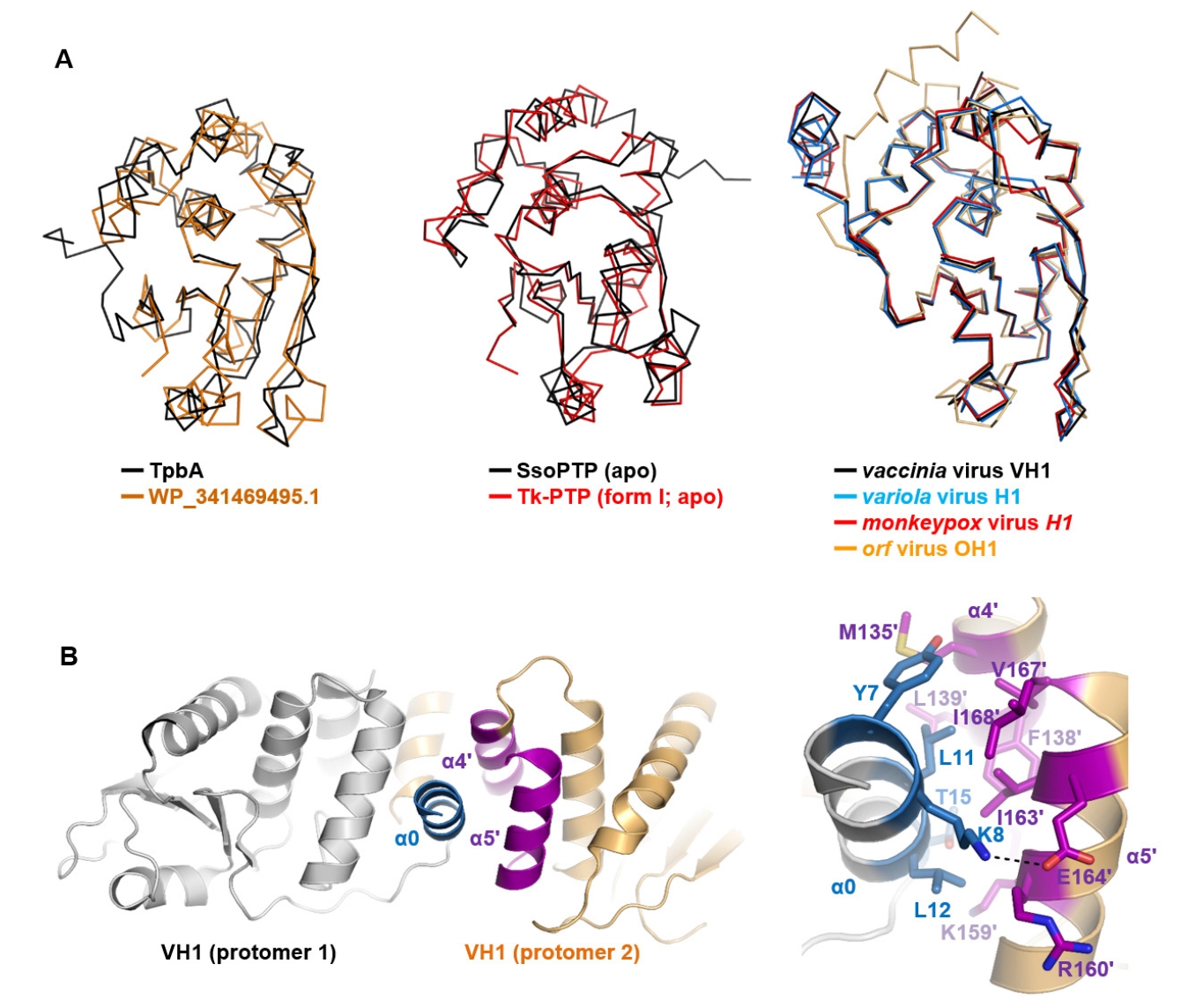
Fig. 4.
| Protein | Alias | PDB code | References |
|---|---|---|---|
| DUSP1 | MKP-1, VH1 | 6APX, 6D65, 6D66, 6D67 | |
| DUSP2 | PAC-1 | 1M3G |
|
| DUSP4 | MKP-2, VH2 | 3EZZ | |
| DUSP5 | VH3 | 2G6Z | |
| DUSP6 | MKP-3, Pyst1 | 1MKP | |
| DUSP7 | MKP-X, Pyst2 | 4Y2E | |
| DUSP8 | VH5 | 4JMK | |
| DUSP9 | MKP-4, Pyst3 | 2HXP, 3LJ8 | |
| DUSP10 | MKP-5 | 1ZZW, 2OUD, 6MC1, 7U4O, 7U4R, 7UMU, 7UMV, 7UN0, 7UN4, 7Y4B, 7Y4C, 7Y4D, 7Y4E | |
| DUSP14 | MKP-6 | 2WGP | |
| DUSP16 | MKP-7 | 4YR8 |
| Protein | Alias | PDB code | References |
|---|---|---|---|
| DUSP3 | VHR | 1J4X, 1VHR, 3F81, 8TK2, 8TK3, 8TK4, 8TK5, 8TK6, 9DJ9 | |
| DUSP11 | PIR1 | 4JMJ, 4MBB, 4NYH | |
| DUSP12 | YVH1 | 4JNB, 4KI9 | |
| DUSP13a | BEDP | 5XJV | |
| DUSP13b | SKRP4, TMDP | 2GWO, 2PQ5 | |
| DUSP15 | VHY | 1YZ4 | |
| DUSP18 | DUSP20 | 2ESB | |
| DUSP19 | DUSP17, SKRP1 | 3S4E, 4D3P, 4D3Q, 4D3R | |
| DUSP21 | - | (none) | (none) |
| DUSP22 | JSP1, VHX | 1WRM, 4WOH, 6L1S, 6LMY, 6LOT, 6LOU, 6LVQ, 7C8S | |
| DUSP23a | VHZ, DUSP25 | 2IMG, 4ERC | |
| DUSP23b | PTPMT1, MOSP | (none) | (none) |
| DUSP26 | DUSP24, SKRP3 | 2E0T, 4B04, 4HRF, 5GTJ | |
| DUSP28 | VHP | 5Y15, 5Y16 | |
| DUSP29 | DUSP27. DUPD1 | 2Y96 |
| Classification | Species | Protein | PDB code | References |
|---|---|---|---|---|
| Bacteria | P. aeruginosa | TpbA | 2M3V |
|
| Archaea | T. kodakarensis | Tk-PTP | 5Z59, 5Z5A, 5Z5B | |
| S. solfataricus | SsoPTP | 2DXP, 2I6I, 2I6J, 2I6M, 2I6O, 2I6P, 7MPC, PMPD, | ||
| Virus | Variola virus | H1 | 2P4D | |
| Vaccinia virus | VH1 | 2Q05, 2RF6, 3CM3 | ||
| Monkeypox virus | H1 | 8GZ4 | ||
| Orf virus | OH1 | 5NCR |
| Protein | PDB code | Residues | Intramolecular C−C contacts (< 4.5 Å) |
|---|---|---|---|
| DUSP3 | 1J4X | 8–185 | 542 (3.04 / 1 residue) |
| DUSP28 | 5Y15 | 12–159 | 439 (2.97 / 1 residue) |
| Tk-PTP | 5Z5A | 1–147 | 632 (4.30 / 1 residue) |
| SsoPTP | 2I6I | 1–161 | 633 (3.93 / 1 residue) |
Structure determined by nuclear magnetic resonance; all other structures were determined by X-ray crystallography.
All the structures were determined by X-ray crystallography. The word “none” indicates structures that have not yet been elucidated.
Structure determined by nuclear magnetic resonance; all other structures were determined by X-ray crystallography.
Table 1.
Table 2.
Table 3.
Table 4.
TOP

