- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 64(3); 2026 > Review
-
Review
Structural perspectives on clinical β-lactamase inhibitors: From mechanism to resistance - Soo-Bong Park1,†, Myeong-Yeon Kim1,†, Sun-Shin Cha1,2,3,*
-
Journal of Microbiology 2026;64(3):e2510019.
DOI: https://doi.org/10.71150/jm.2510019
Published online: March 19, 2026
1Department of Chemistry and Nanoscience, Ewha Womans University, Seoul 03760, Republic of Korea
2Graduate Program in Innovative Biomaterials Convergence, Ewha Womans University, Seoul 03760, Republic of Korea
3R&D Division, TODD PHARM CO. LTD., Seoul 03760, Republic of Korea
- *Correspondence Sun-Shin Cha chajung@ewha.ac.kr
- †These authors contributed equally to this work.
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- Introduction
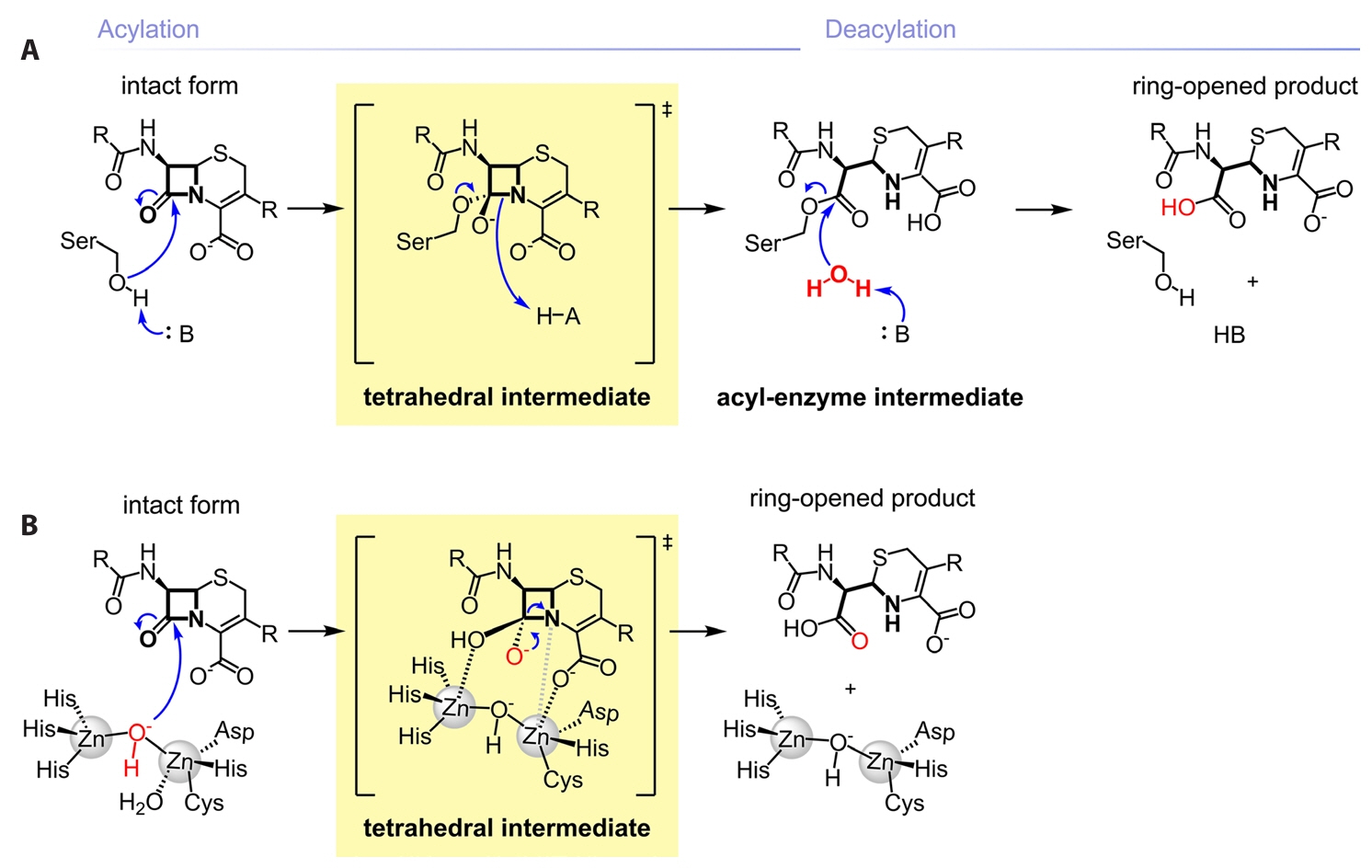
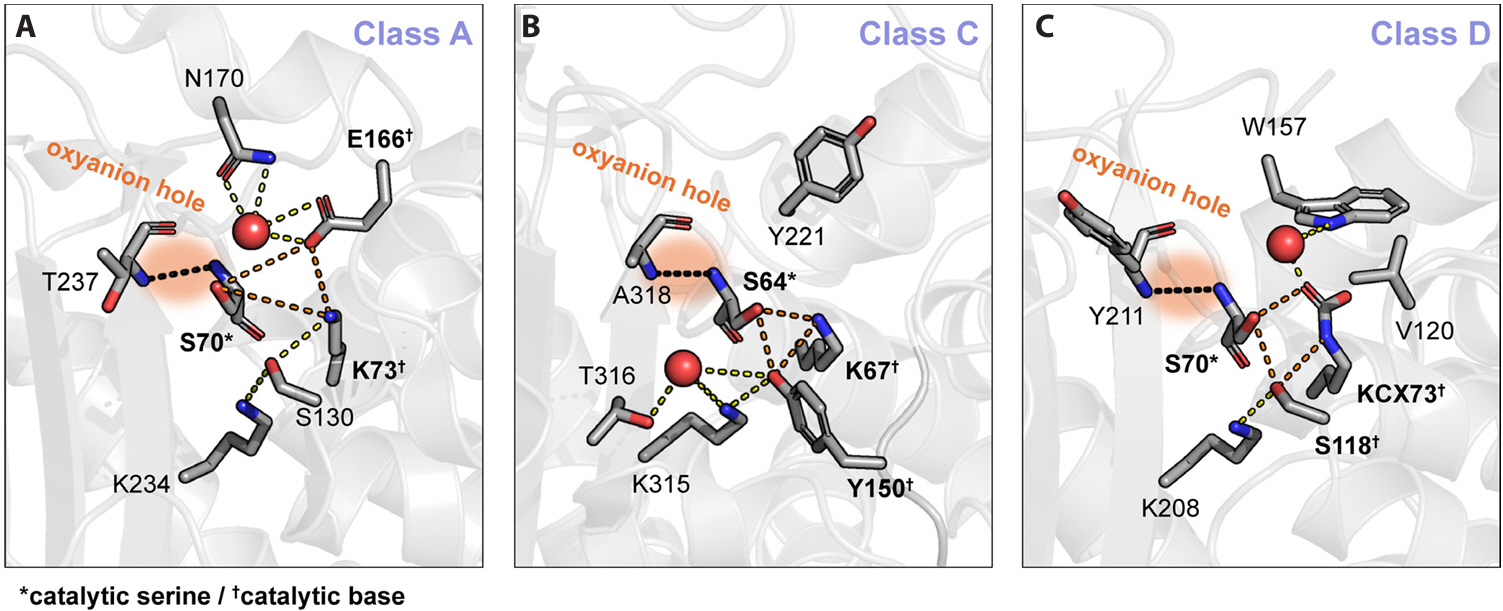
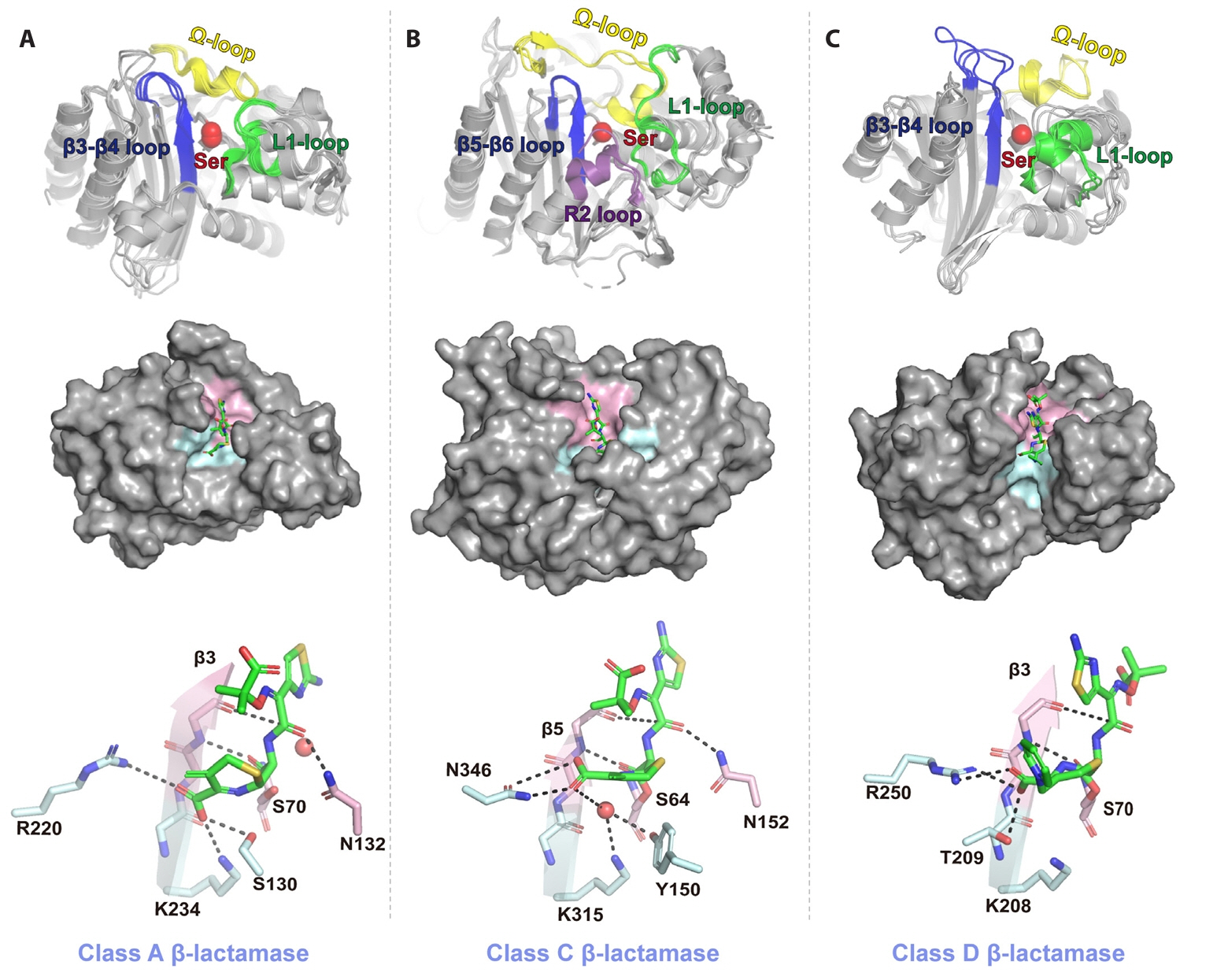
- Mechanistic and Structural Diversity of β-lactamases
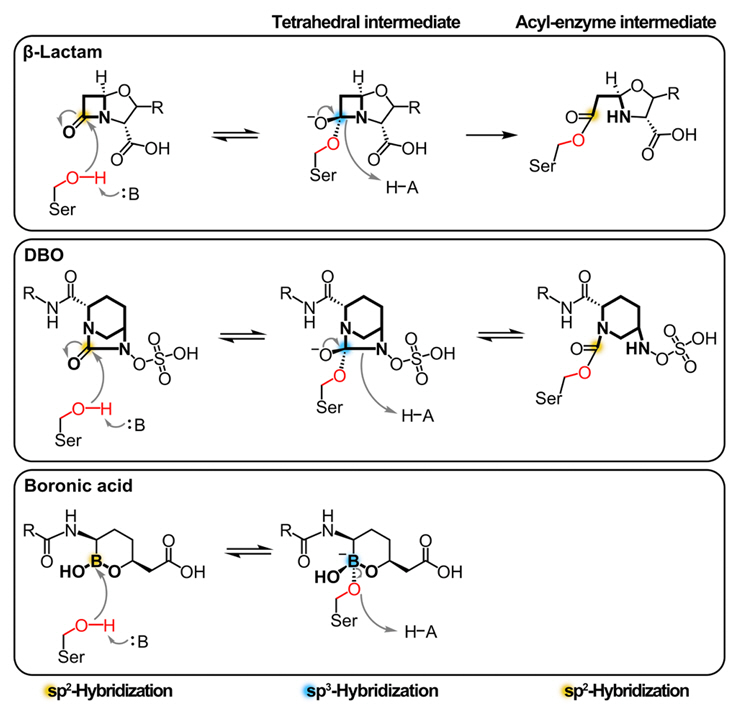
- Structural Overview of Mechanisms and Limitations in FDA-Approved BLIs
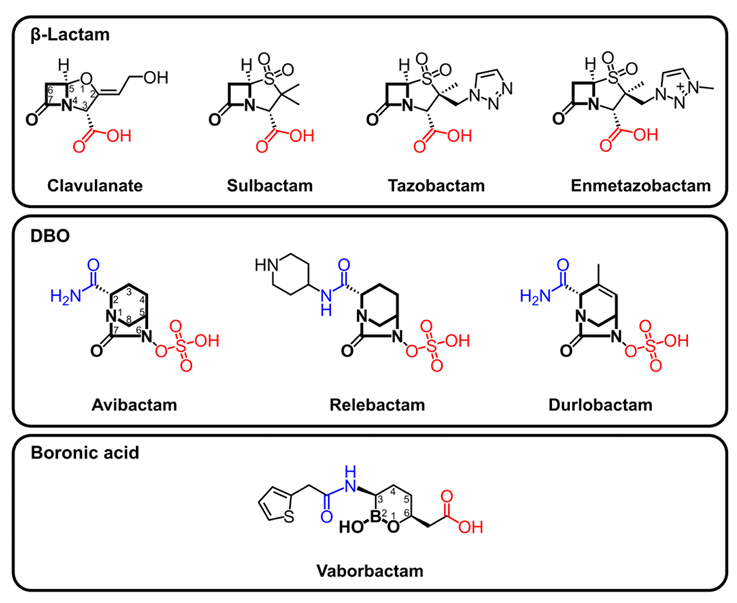
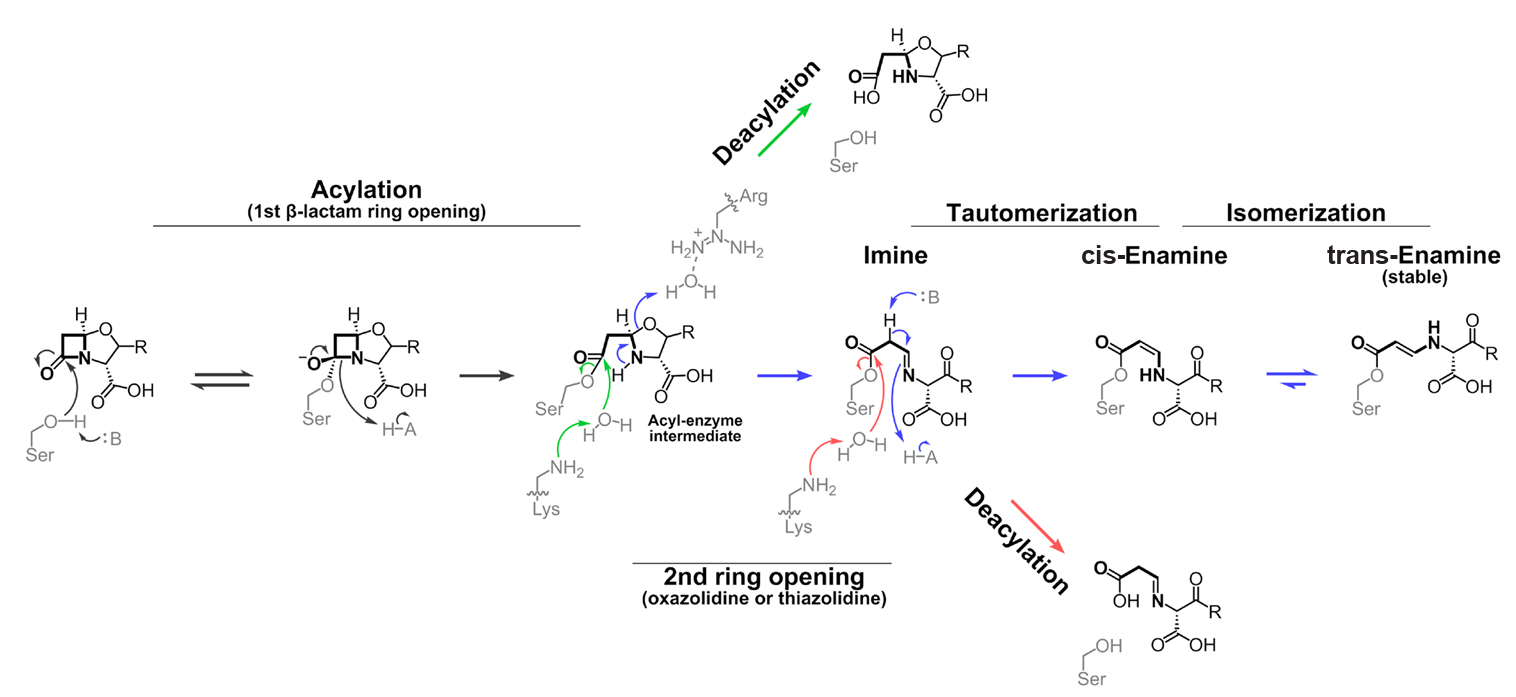
- β-Lactam-Based BLIs
- DBO-based BLIs
- Boronic Acid-based BLIs
- Expanding the Therapeutic Potential of BLIs through PBP Inhibition
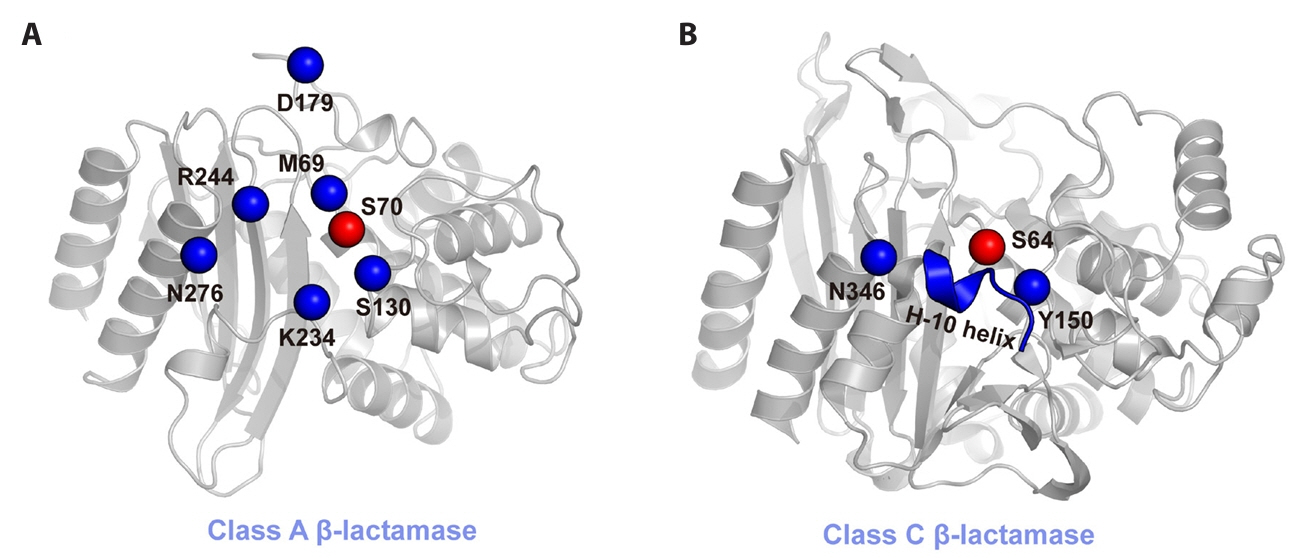
- Clinical Emergence of BLI-Resistant Variants
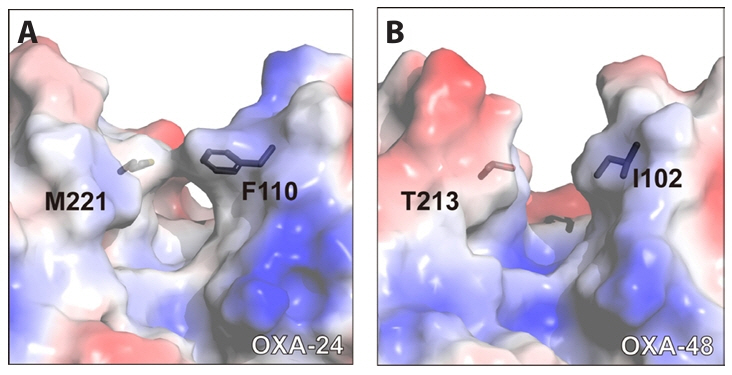
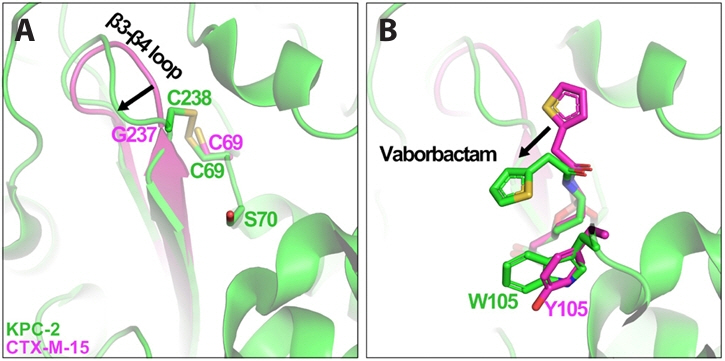
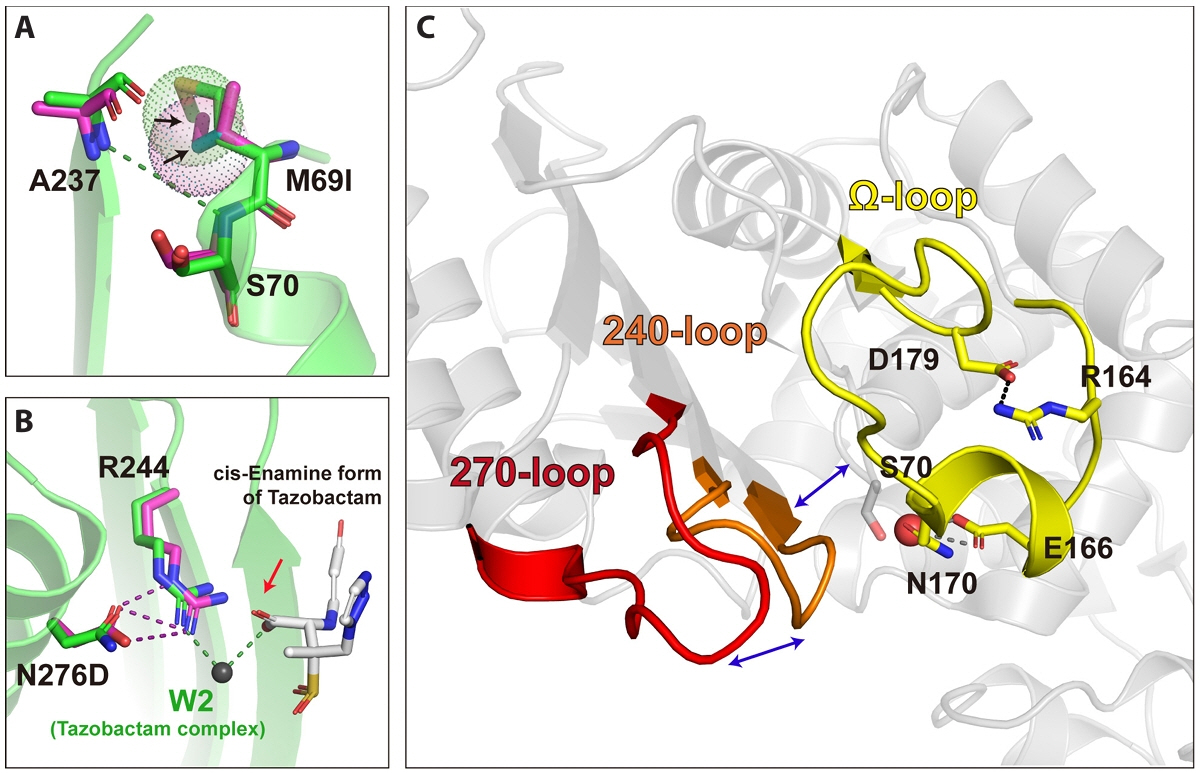
- Structural Mechanisms of Resistance to β-Lactam-Based Inhibitors
- Structural Mechanisms of Resistance to DBO-Based Inhibitors
- Vaborbactam Resistance Arises without Enzyme Modification
- Discussion
- Notes
- References
ABSTRACT
- β-Lactam antibiotics marked the beginning of an era of effective and safe treatment for bacterial infections and remain the most widely prescribed antibacterial agents today. However, the emergence of antibiotic-resistant bacteria threatens a return to the pre-antibiotic era. In particular, bacterial expression of β-lactamases inactivating β-lactam antibiotics presents a challenge in antimicrobial therapy. While inhibitors against β-lactamases have been developed to protect the therapeutic efficacy of β-lactam antibiotics, the clinical use of β-lactamase inhibitors is constrained due to their limited inhibition spectrum and the emergence of inhibitor-resistant β-lactamase variants. As an effort to tackle this issue, here we reviewed the structural and mechanistic features of β-lactamases and their FDA-approved inhibitors. Moreover, mutations in clinically isolated β-lactamases that confer resistance against their inhibitors are compiled. The comprehensive overview offered in this review aims to support and stimulate the design of next-generation β-lactamase inhibitors for combating β-lactamase-mediated antibiotic resistance.
Introduction
Mechanistic and Structural Diversity of β-lactamases
Structural Overview of Mechanisms and Limitations in FDA-Approved BLIs
β-Lactam-Based BLIs
DBO-based BLIs
Boronic Acid-based BLIs
Expanding the Therapeutic Potential of BLIs through PBP Inhibition
Clinical Emergence of BLI-Resistant Variants
Structural Mechanisms of Resistance to β-Lactam-Based Inhibitors
Structural Mechanisms of Resistance to DBO-Based Inhibitors
Vaborbactam Resistance Arises without Enzyme Modification
Discussion
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (grant No. RS-2022-NR070574, RS-2022-NR070833, and RS-2024-00352229) and Korea Institute of Ocean Science and Technology (grant No. PEA0025).
Conflict of Interest
The authors have no conflict of interest to report.
Ethical Statements
This study did not involve human participants or animals, and therefore ethical approval was not required.
| Core structure | Inhibitor | Partner β-lactam | Formulation (Year of FDA approval) | Inhibition profile of the inhibitor† | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Class A | Class C | Class D | Class B | |||||||||
| TEM | SHV | CTX-M | KPC | AmpC | CMY | OXA-23 | OXA-48 | MBL‡ | ||||
| β-Lactam | Clavulanic acid | Amoxicillin | Augmentin (1984) | O | O | O | X | X | X | X | X | X |
| Sulbactam | Ampicillin | Unasyn (1986) | O | △ | △ | X | X | X | X | X | X | |
| Tazobactam | Piperacillin | Zosyn (1993) | O | O | O | X | X | X | X | X | X | |
| Ceftolozane | Zerbaxa (2014) | |||||||||||
| Enmetazobactam | Cefepime | Exblifep (2024) | O | O | O | X | X | X | X | X | X | |
| DBO* | Avibactam | Ceftazidime | Avycaz (2015) | O | O | O | O | O | O | X | O | X |
| Aztreonam | Emblaveo (2025) | |||||||||||
| Relebactam | Imipenem and cilastatin | Recarbrio (2019) | O | O | O | O | O | O | X | X | X | |
| Durlobactam | Sulbactam | Xacduro (2023) | O | O | O | O | O | O | △ | O | X | |
| Boronic acid | Vaborbactam | Meropenem | Vabomere(2017) | O | O | O | O | O | O | X | X | X |
- Alsenani TA, Viviani SL, Kumar V, Taracila MA, Bethel CR, et al. 2022. Structural characterization of the D179N and D179Y variants of KPC-2 β-lactamase: Ω-loop destabilization as a mechanism of resistance to ceftazidime-avibactam. Antimicrob Agents Chemother. 66: e0241421. ArticlePubMedLink
- Alsenani TA, Viviani SL, Papp-Wallace KM, Bonomo RA, van den Akker F. 2023. Exploring avibactam and relebactam inhibition of Klebsiella pneumoniae carbapenemase D179N variant: role of the Ω loop-held deacylation water. Antimicrob Agents Chemother. 67: e0035023. ArticlePubMedLink
- Ambler RP. 1980. The structure of β-lactamases. Philos Trans R Soc Lond B Biol Sci. 289: 321–331. ArticlePubMedPDF
- Ambler RP, Coulson AF, Frere JM, Ghuysen JM, Joris B, et al. 1991. A standard numbering scheme for the class A β-lactamases. Biochem J. 276: 269–270. ArticlePubMedPMCPDF
- Asli A, Brouillette E, Krause KM, Nichols WW, Malouin F. 2016. Distinctive binding of avibactam to penicillin-binding proteins of Gram-negative and Gram-positive bacteria. Antimicrob Agents Chemother. 60: 752–756. ArticlePubMedPMCLink
- Barnes MD, Kumar V, Bethel CR, Moussa SH, O'Donnell J, et al. 2019. Targeting multidrug-resistant Acinetobacter spp.: sulbactam and the diazabicyclooctenone β-lactamase inhibitor ETX2514 as a novel therapeutic agent. mBio. 10: e00159-19.ArticlePubMedPMCLink
- Bonomo RA, Dawes CG, Knox JR, Shlaes DM. 1995. Complementary roles of mutations at positions 69 and 242 in a class A β-lactamase. Biochim Biophys Acta. 1247: 113–120. ArticlePubMed
- Bonomo RA, Liu J, Chen Y, Ng L, Hujer AM, et al. 2001. Inactivation of CMY-2 β-lactamase by tazobactam: initial mass spectroscopic characterization. Biochim Biophys Acta. 1547: 196–205. ArticlePubMed
- Brown RP, Aplin RT, Schofield CJ. 1996. Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry. 35: 12421–12432. ArticlePubMed
- Bush K. 2018. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother. 62: e01076-18.ArticlePubMedPMCLink
- Bush K, Bradford PA. 2016. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med. 6: a025247.ArticlePubMedPMC
- Bush K, Bradford PA. 2020. Epidemiology of β-lactamase-producing pathogens. Clin Microbiol Rev. 33: e00046-19.ArticlePubMedPMCLink
- Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 54: 969–976. ArticlePubMedLink
- Bush K, Macalintal C, Rasmussen BA, Lee VJ, Yang Y. 1993. Kinetic interactions of tazobactam with β-lactamases from all major structural classes. Antimicrob Agents Chemother. 37: 851–858. ArticlePubMedPMCLink
- Canica MM, Caroff N, Barthelemy M, Labia R, Krishnamoorthy R, et al. 1998. Phenotypic study of resistance of β-lactamase-inhibitor-resistant TEM enzymes which differ by naturally occurring variations and by site-directed substitution at Asp276. Antimicrob Agents Chemother. 42: 1323–1328. ArticlePubMedPMCLink
- Canton R, Morosini MI, de la Maza OM, de la Pedrosa EG. 2008. IRT and CMT β-lactamases and inhibitor resistance. Clin Microbiol Infect. 14: 53–62. ArticlePubMed
- Chaibi EB, Peduzzi J, Farzaneh S, Barthelemy M, Sirot D, et al. 1998. Clinical inhibitor-resistant mutants of the β-lactamase TEM-1 at amino-acid position 69: kinetic analysis and molecular modelling. Biochim Biophys Acta. 1382: 38–46. ArticlePubMed
- Chen CC, Herzberg O. 1992. Inhibition of β-lactamase by clavulanate: trapped intermediates in cryocrystallographic studies. J Mol Biol. 224: 1103–1113. ArticlePubMedPMC
- Chen AY, Thomas PW, Stewart AC, Bergstrom A, Cheng Z, et al. 2017. Dipicolinic acid derivatives as inhibitors of New Delhi metallo-β-lactamase-1. J Med Chem. 60: 7267–7283. ArticlePubMed
- Choi H, Paton RS, Park H, Schofield CJ. 2016. Investigations on recyclisation and hydrolysis in avibactam-mediated serine β-lactamase inhibition. Org Biomol Chem. 14: 4116–4128. ArticlePubMedPMC
- Coleman K. 2011. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr Opin Microbiol. 14: 550–555. ArticlePubMed
- Compain F, Arthur M. 2017. Impaired inhibition by avibactam and resistance to the ceftazidime-avibactam combination due to the D179Y substitution in the KPC-2 β-lactamase. Antimicrob Agents Chemother. 61: e00451-17.ArticlePubMedPMCLink
- Compain F, Debray A, Adjadj P, Dorchene D, Arthur M. 2020. Ceftazidime-avibactam resistance mediated by the N346Y substitution in various AmpC β-lactamases. Antimicrob Agents Chemother. 64: e02311-19.ArticlePubMedLink
- Das CK, Nair NN. 2018. Molecular insights into avibactam-mediated class C β-lactamase inhibition: competition between reverse acylation and hydrolysis through desulfation. Phys Chem Chem Phys. 20: 14482–14490. Article
- De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, et al. 2020. Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev. 33: e00181-19.ArticlePubMedPMC
- Diaz DB, Yudin AK. 2017. The versatility of boron in biological target engagement. Nat Chem. 9: 731–742. ArticlePubMedPDF
- Drawz SM, Bethel CR, Hujer KM, Hurless KN, Distler AM, et al. 2009. The role of a second-shell residue in modifying substrate and inhibitor interactions in the SHV β-lactamase: a study of Ambler position Asn276. Biochemistry. 48: 4557–4566. ArticlePubMed
- Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 23: 160–201. ArticlePubMedPMCLink
- Dulyayangkul P, Wan Nur Ismah WAK, Douglas EJA, Avison MB. 2020. Mutation of kvrA causes OmpK35 and OmpK36 porin downregulation and reduced meropenem-vaborbactam susceptibility in KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 64: e02208-19.ArticlePubMedPMCLink
- Durand-Reville TF, Guler S, Comita-Prevoir J, Chen B, Bifulco N, et al. 2017. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat Microbiol. 2: 17104.ArticlePubMed
- Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, et al. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci USA. 109: 11663–11668. ArticlePubMedPMC
- Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, et al. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem. 288: 27960–27971. ArticlePubMedPMC
- Elings W, Chikunova A, van Zanten DB, Drenth R, Ahmad MUD, et al. 2021. Two β-lactamase variants with reduced clavulanic acid inhibition display different millisecond dynamics. Antimicrob Agents Chemother. 65: e02628-20.ArticlePubMedPMCLink
- Finlay J, Miller L, Poupard JA. 2003. A review of the antimicrobial activity of clavulanate. J Antimicrob Chemother. 52: 18–23. ArticlePubMed
- Giakkoupi P, Miriagou V, Gazouli M, Tzelepi E, Legakis NJ, et al. 1998a. Properties of mutant SHV-5 β-lactamases constructed by substitution of isoleucine or valine for methionine at position 69. Antimicrob Agents Chemother. 42: 1281–1283. ArticleLink
- Giakkoupi P, Tzelepi E, Legakis NJ, Tzouvelekis LS. 1998b. Substitution of Arg-244 by Cys or Ser in SHV-1 and SHV-5 β-lactamases confers resistance to mechanism-based inhibitors and reduces catalytic efficiency of the enzymes. FEMS Microbiol Lett. 160: 49–54. Article
- Gonzalez-Bello C, Rodriguez D, Pernas M, Rodriguez A, Colchon E. 2020. β-Lactamase inhibitors to restore the efficacy of antibiotics against superbugs. J Med Chem. 63: 1859–1881. ArticlePubMedLink
- Hall BG, Barlow M. 2003. Structure-based phylogenies of the serine β-lactamases. J Mol Evol. 57: 255–260. ArticlePubMedPDF
- Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, et al. 2015. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem. 58: 3682–3692. ArticlePubMed
- Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, et al. 2003. Understanding resistance to β-lactams and β-lactamase inhibitors in the SHV β-lactamase: lessons from the mutagenesis of Ser-130. J Biol Chem. 278: 52724–52729. ArticlePubMed
- Hinchliffe P, Tooke CL, Bethel CR, Wang B, Arthur C, et al. 2022. Penicillanic acid sulfones inactivate the extended-spectrum β-lactamase CTX-M-15 through formation of a serine-lysine cross-link: an alternative mechanism of β-lactamase inhibition. mBio. 13: e01793-21.ArticleLink
- Hoff JF, Goudar KE, Calvopina K, Beer M, Hinchliffe P, et al. 2025. Electrostatic interactions influence diazabicyclooctane inhibitor potency against OXA-48-like β-lactamases. RSC Med Chem. 16: 5441–5455. ArticlePubMedPMC
- Hugonnet JE, Blanchard JS. 2007. Irreversible inhibition of the Mycobacterium tuberculosis β-lactamase by clavulanate. Biochemistry. 46: 11998–12004. ArticlePubMed
- Imtiaz U, Billings E, Knox JR, Manavathu EK, Lerner SA, et al. 1993. Inactivation of class A β-lactamases by clavulanic acid: the role of arginine-244 in a proposed nonconcerted sequence of events. J Am Chem Soc. 115: 4435–4442. Article
- Jeong BG, Na JH, Bae DW, Park SB, Lee HS, et al. 2021. Crystal structure of AmpC BER and molecular docking lead to the discovery of broad inhibition activities of halisulfates against β-lactamases. Comput Struct Biotechnol J. 19: 145–152. ArticlePubMed
- Ji Z, Kozuch J, Mathews II, Diercks CS, Shamsudin Y, et al. 2022. Protein electric fields enable faster and longer-lasting covalent inhibition of β-lactamases. J Am Chem Soc. 144: 20947–20954. ArticlePubMedPMCLink
- Kalp M, Totir MA, Buynak JD, Carey PR. 2009. Different intermediate populations formed by tazobactam, sulbactam, and clavulanate reacting with SHV-1 β-lactamases: raman crystallographic evidence. J Am Chem Soc. 131: 2338–2347. ArticlePubMedPMC
- Kim MK, An YJ, Na JH, Seol JH, Ryu JY, et al. 2017. Structural and mechanistic insights into the inhibition of class C β-lactamases through the adenylylation of the nucleophilic serine. J Antimicrob Chemother. 72: 735–743. ArticlePubMed
- Kim JY, Jung HI, An YJ, Lee JH, Kim SJ, et al. 2006. Structural basis for the extended substrate spectrum of CMY-10, a plasmid-encoded class C β-lactamase. Mol Microbiol. 60: 907–916. ArticlePubMed
- Kim D, Kim S, Kwon Y, Kim Y, Park H, et al. 2023. Structural insights for β-lactam antibiotics. Biomol Ther. 31: 141–147. Article
- Klein EY, Impalli I, Poleon S, Denoel P, Cipriano M, et al. 2024. Global trends in antibiotic consumption during 2016–2023 and future projections through 2030. Proc Natl Acad Sci USA. 121: e2411919121. ArticlePubMedPMC
- Krajnc A, Lang PA, Panduwawala TD, Brem J, Schofield CJ. 2019. Will morphing boron-based inhibitors beat the β-lactamases? Curr Opin Chem Biol. 50: 101–110. ArticlePubMedPMC
- Krishnan NP, Nguyen NQ, Papp-Wallace KM, Bonomo RA, van den Akker F. 2015. Inhibition of Klebsiella β-lactamases (SHV-1 and KPC-2) by avibactam: a structural study. PLoS One. 10: e0136813. ArticlePubMedPMC
- Kumar V, Viviani SL, Ismail J, Agarwal S, Bonomo RA, et al. 2021. Structural analysis of the boronic acid β-lactamase inhibitor vaborbactam binding to Pseudomonas aeruginosa penicillin-binding protein 3. PLoS One. 16: e0258359. ArticlePubMedPMC
- Lahiri SD, Giacobbe RA, Johnstone MR, Alm RA. 2014a. Activity of avibactam against Enterobacter cloacae producing an extended-spectrum class C β-lactamase enzyme. J Antimicrob Chemother. 69: 2942–2946. Article
- Lahiri SD, Johnstone MR, Ross PL, McLaughlin RE, Olivier NB, et al. 2014b. Avibactam and class C β-lactamases: mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob Agents Chemother. 58: 5704–5713. ArticleLink
- Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, et al. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother. 57: 2496–2505. ArticlePubMedPMCLink
- Lahiri SD, Mangani S, Jahic H, Benvenuti M, Durand-Reville TF, et al. 2015. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: a structure-guided study of OXA-24 and OXA-48. ACS Chem Biol. 10: 591–600. ArticlePubMed
- Lang PA, de Munnik M, Oluwole AO, Claridge TDW, Robinson CV, et al. 2024. How clavulanic acid inhibits serine β-lactamases. Chembiochem. 25: e202400280. ArticlePubMed
- Lang PA, Leissing TM, Page MGP, Schofield CJ, Brem J. 2021. Structural investigations of the inhibition of Escherichia coli AmpC β-lactamase by diazabicyclooctanes. Antimicrob Agents Chemother. 65: e02073-20.ArticlePubMedPMCLink
- Lang PA, Raj R, Tumber A, Lohans CT, Rabe P, et al. 2022. Studies on enmetazobactam clarify mechanisms of widely used β-lactamase inhibitors. Proc Natl Acad Sci USA. 119: e2117310119. ArticlePubMedPMC
- Lee JH, Jeong SH, Cha SS, Lee SH. 2009. New disturbing trend in antimicrobial resistance of Gram-negative pathogens. PLoS Pathog. 5: e1000221. ArticlePubMedPMC
- Lee H, Park H, Kwak K, Lee CE, Yun J, et al. 2025. Structural comparison of substrate-binding pockets of serine β-lactamases in classes A, C, and D. J Enzyme Inhib Med Chem. 40: 2435365.ArticlePubMed
- Li GB, Abboud MI, Brem J, Someya H, Lohans CT, et al. 2017. NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors. Chem Sci. 8: 928–937. ArticlePubMed
- Li R, Liao JM, Gu CR, Wang YT, Chen CL. 2011. Theoretical investigation on reaction of sulbactam with wild-type SHV-1 β-lactamase: acylation, tautomerization, and deacylation. J Phys Chem B. 115: 10298–10310. ArticlePubMed
- Liu B, Trout REL, Chu GH, McGarry D, Jackson RW, et al. 2020. Discovery of taniborbactam (VNRX-5133): a broad-spectrum serine- and metallo-β-lactamase inhibitor for carbapenem-resistant bacterial infections. J Med Chem. 63: 2789–2801. ArticlePubMed
- Lizana I, Pecchi G, Uribe EA, Delgado EJ. 2022. A rationale for the unlike potency of avibactam and ETX2514 against OXA-24 β-lactamase. Arch Biochem Biophys. 727: 109343.ArticlePubMed
- Lobkovsky E, Moews PC, Liu H, Zhao H, Frere JM, et al. 1993. Evolution of an enzyme activity: crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc Natl Acad Sci USA. 90: 11257–11261. ArticlePubMedPMC
- Lomovskaya O, Sun D, Rubio-Aparicio D, Nelson K, Tsivkovski R, et al. 2017. Vaborbactam: spectrum of β-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob Agents Chemother. 61: e01443-17.ArticlePubMedPMC
- Marciano DC, Brown NG, Palzkill T. 2009. Analysis of the plasticity of location of the Arg244 positive charge within the active site of the TEM-1 β-lactamase. Protein Sci. 18: 2080–2089. ArticlePubMedPMCLink
- Mehta SC, Furey IM, Pemberton OA, Boragine DM, Chen Y, et al. 2021. KPC-2 β-lactamase enables carbapenem antibiotic resistance through fast deacylation of the covalent intermediate. J Biol Chem. 296: 100155.ArticlePubMed
- Meletis G. 2016. Carbapenem resistance: overview of the problem and future perspectives. Ther Adv Infect Dis. 3: 15–21. ArticlePubMedPMCLink
- Mojica MF, Rossi MA, Vila AJ, Bonomo RA. 2022. The urgent need for metallo-β-lactamase inhibitors: an unattended global threat. Lancet Infect Dis. 22: e28–e34. ArticlePubMed
- Monnaie D, Frere JM. 1993. Interaction of clavulanate with class C β-lactamases. FEBS Lett. 334: 269–271. ArticlePubMedLink
- Mora-Ochomogo M, Lohans CT. 2021. β-Lactam antibiotic targets and resistance mechanisms: from covalent inhibitors to substrates. RSC Med Chem. 12: 1623–1639. ArticlePubMedPMC
- Moya B, Barcelo IM, Bhagwat S, Patel M, Bou G, et al. 2017. Potent β-lactam enhancer activity of zidebactam and WCK 5153 against Acinetobacter baumannii, including carbapenemase-producing clinical isolates. Antimicrob Agents Chemother. 61: e01238-17.ArticlePubMedPMCLink
- Na JH, An YJ, Cha SS. 2017. GMP and IMP are competitive inhibitors of CMY-10, an extended-spectrum class C β-lactamase. Antimicrob Agents Chemother. 61: e00098-17.ArticlePubMedPMCLink
- Na JH, Cha SS. 2016. Structural basis for the extended substrate spectrum of AmpC BER and structure-guided discovery of the inhibition activity of citrate against the class C β-lactamases AmpC BER and CMY-10. Acta Crystallogr D Struct Biol. 72: 976–985. ArticlePubMed
- Na JH, Lee TH, Park SB, Kim MK, Jeong BG, et al. 2018. In vitro and in vivo inhibitory activity of NADPH against the AmpC BER class C β-lactamase. Front Cell Infect Microbiol. 8: 441.ArticlePubMedPMC
- Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, et al. 2017. β-lactamase database (BLDB): structure and function. J Enzyme Inhib Med Chem. 32: 917–919. ArticlePubMedPMC
- Papp-Wallace KM. 2019. The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin Pharmacother. 20: 2169–2184. ArticlePubMedPMC
- Papp-Wallace KM, McLeod SM, Miller AA. 2023. Durlobactam, a broad-spectrum serine β-lactamase inhibitor, restores sulbactam activity against Acinetobacter species. Clin Infect Dis. 76: S194–S201. ArticlePubMedPMCPDF
- Papp-Wallace KM, Nguyen NQ, Jacobs MR, Bethel CR, Barnes MD, et al. 2018. Strategic approaches to overcome resistance against Gram-negative pathogens using β-lactamase inhibitors and β-lactam enhancers: activity of three novel diazabicyclooctanes WCK 5153, zidebactam (WCK 5107), and WCK 4234. J Med Chem. 61: 4067–4086. ArticlePubMedPMC
- Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, Bonomo RA. 2012. Understanding the molecular determinants of substrate and inhibitor specificities in the Carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob Agents Chemother. 56: 4428–4438. ArticlePubMedPMCLink
- Payne DJ, Cramp R, Winstanley DJ, Knowles DJ. 1994. Comparative activities of clavulanic acid, sulbactam, and tazobactam against clinically important β-lactamases. Antimicrob Agents Chemother. 38: 767–772. ArticlePubMedPMCLink
- Pemberton OA, Tsivkovski R, Totrov M, Lomovskaya O, Chen Y. 2020. Structural basis and binding kinetics of vaborbactam in class A β-lactamase inhibition. Antimicrob Agents Chemother. 64: e00398-20.ArticlePubMedPMCLink
- Philippon A, Arlet G, Labia R, Iorga BI. 2022. Class C β-lactamases: molecular characteristics. Clin Microbiol Rev. 35: e0015021. ArticlePubMedLink
- Power P, Mercuri P, Herman R, Kerff F, Gutkind G, et al. 2012. Novel fragments of clavulanate observed in the structure of the class A β-lactamase from Bacillus licheniformis BS3. J Antimicrob Chemother. 67: 2379–2387. ArticlePubMed
- Reading C, Cole M. 1977. Clavulanic acid: a β-lactamase-inhibiting β-lactam from Streptomyces clavuligerus. Antimicrob Agents Chemother. 11: 852–857. ArticlePubMedPMCLink
- Ruggiero M, Kerff F, Herman R, Sapunaric F, Galleni M, et al. 2014. Crystal structure of the extended-spectrum β-lactamase PER-2 and insights into the role of specific residues in the interaction with β-lactams and β-lactamase inhibitors. Antimicrob Agents Chemother. 58: 5994–6002. ArticlePubMedPMCLink
- Russ D, Glaser F, Shaer Tamar E, Yelin I, Baym M, et al. 2020. Escape mutations circumvent a tradeoff between resistance to a β-lactam and resistance to a β-lactamase inhibitor. Nat Commun. 11: 2029.ArticlePubMedPMC
- Shapiro AB. 2017. Kinetics of sulbactam hydrolysis by β-lactamases, and kinetics of β-lactamase inhibition by sulbactam. Antimicrob Agents Chemother. 61: e01612-17.ArticlePubMedPMCLink
- Shapiro AB, Moussa SH, McLeod SM, Durand-Reville T, Miller AA. 2021. Durlobactam, a new diazabicyclooctane β-lactamase inhibitor for the treatment of Acinetobacter infections in combination with sulbactam. Front Microbiol. 12: 709974.ArticlePubMedPMC
- Shields RK, Iovleva A, Kline EG, Kawai A, McElheny CL, et al. 2020. Clinical evolution of AmpC-mediated ceftazidime-avibactam and cefiderocol resistance in Enterobacter cloacae complex following exposure to cefepime. Clin Infect Dis. 71: 2713–2716. ArticlePubMedPMCPDF
- Soeung V, Lu S, Hu L, Judge A, Sankaran B, et al. 2020. A drug-resistant β-lactamase variant changes the conformation of its active-site proton shuttle to alter substrate specificity and inhibitor potency. J Biol Chem. 295: 18239–18255. ArticlePubMed
- Stewart NK, Smith CA, Antunes NT, Toth M, Vakulenko SB. 2019. Role of the hydrophobic bridge in the carbapenemase activity of class D β-lactamases. Antimicrob Agents Chemother. 63: e02191-18.ArticlePubMedPMCLink
- Stewart NK, Toth M, Alqurafi MA, Chai W, Nguyen TQ, et al. 2022. C6 hydroxymethyl-substituted carbapenem MA-1-206 inhibits the major Acinetobacter baumannii carbapenemase OXA-23 by impeding deacylation. mBio. 13: e0036722. ArticlePubMedLink
- Strynadka NC, Jensen SE, Johns K, Blanchard H, Page M, et al. 1994. Structural and kinetic characterization of a β-lactamase-inhibitor protein. Nature. 368: 657–660. ArticlePubMedPDF
- Sun T, Bethel CR, Bonomo RA, Knox JR. 2004. Inhibitor-resistant class A β-lactamases: consequences of the Ser130-to-Gly mutation seen in apo and tazobactam structures of the SHV-1 variant. Biochemistry. 43: 14111–14117. ArticlePubMed
- Sun Z, Lin H, Hu L, Neetu N, Sankaran B, et al. 2024. Klebsiella pneumoniae carbapenemase variant 44 acquires ceftazidime-avibactam resistance by altering the conformation of active-site loops. J Biol Chem. 300: 105493.ArticlePubMed
- Sun D, Rubio-Aparicio D, Nelson K, Dudley MN, Lomovskaya O. 2017. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 61: e01694-17.ArticlePubMedPMCLink
- Swaren P, Golemi D, Cabantous S, Bulychev A, Maveyraud L, et al. 1999. X-ray structure of the Asn276Asp variant of the Escherichia coli TEM-1 β-lactamase: direct observation of electrostatic modulation in resistance to inactivation by clavulanic acid. Biochemistry. 38: 9570–9576. ArticlePubMed
- Tehrani K, Martin NI. 2018. β-lactam/β-lactamase inhibitor combinations: an update. MedChemComm. 9: 1439–1456. ArticlePubMedPMC
- Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, et al. 2005. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM β-lactamase. Biochemistry. 44: 9330–9338. ArticlePubMed
- Thomson JM, Distler AM, Prati F, Bonomo RA. 2006. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244: understanding unique properties of inhibitor resistance. J Biol Chem. 281: 26734–26744. ArticlePubMed
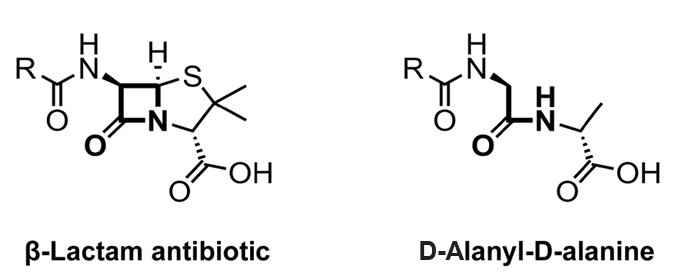
- Tipper DJ, Strominger JL. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc Natl Acad Sci USA. 54: 1133–1141. ArticlePubMedPMC
- Tondi D, Venturelli A, Bonnet R, Pozzi C, Shoichet BK, et al. 2014. Targeting class A and C serine β-lactamases with a broad-spectrum boronic acid derivative. J Med Chem. 57: 5449–5458. ArticlePubMedPMC
- Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, et al. 2019a. β-Lactamases and β-lactamase inhibitors in the 21st century. J Mol Biol. 431: 3472–3500. Article
- Tooke CL, Hinchliffe P, Krajnc A, Mulholland AJ, Brem J, et al. 2020. Cyclic boronates as versatile scaffolds for KPC-2 β-lactamase inhibition. RSC Med Chem. 11: 491–496. ArticlePubMedPMC
- Tooke CL, Hinchliffe P, Lang PA, Mulholland AJ, Brem J, et al. 2019b. Molecular basis of class A β-lactamase inhibition by relebactam. Antimicrob Agents Chemother. 63: e00564-19.ArticleLink
- Toth M, Smith CA, Antunes NT, Stewart NK, Maltz L, et al. 2017. The role of conserved surface hydrophobic residues in the carbapenemase activity of the class D β-lactamases. Acta Crystallogr D Struct Biol. 73: 692–701. ArticlePubMedPMC
- Totir MA, Cha J, Ishiwata A, Wang B, Sheri A, et al. 2008. Why clinically used tazobactam and sulbactam are poor inhibitors of OXA-10 β-lactamase: Raman crystallographic evidence. Biochemistry. 47: 4094–4101. ArticlePubMed
- Tsivkovski R, Lomovskaya O. 2020. Biochemical activity of vaborbactam. Antimicrob Agents Chemother. 64: e01935-19.ArticlePubMedPMCLink
- Usher KC, Blaszczak LC, Weston GS, Shoichet BK, Remington SJ. 1998. Three-dimensional structure of AmpC β-lactamase from Escherichia coli bound to a transition-state analogue: possible implications for the oxyanion hypothesis and for inhibitor design. Biochemistry. 37: 16082–16092. ArticlePubMed
- van den Akker F, Bonomo RA. 2018. Exploring additional dimensions of complexity in inhibitor design for serine β-lactamases: mechanistic and intra- and intermolecular chemistry approaches. Front Microbiol. 9: 622.ArticlePubMedPMC
- Wang YC, Huang SW, Chiang MH, Lee IM, Kuo SC, et al. 2021. In vitro and in vivo activities of imipenem combined with BLI-489 against class D β-lactamase-producing Acinetobacter baumannii. J Antimicrob Chemother. 76: 451–459. ArticlePubMedPDF
- Yocum RR, Waxman DJ, Rasmussen JR, Strominger JL. 1979. Mechanism of penicillin action: penicillin and substrate bind covalently to the same active site serine in two bacterial D-alanine carboxypeptidases. Proc Natl Acad Sci USA. 76: 2730–2734. ArticlePubMedPMC
- Young K, Painter RE, Raghoobar SL, Hairston NN, Racine F, et al. 2019. In vitro studies evaluating the activity of imipenem in combination with relebactam against Pseudomonas aeruginosa. BMC Microbiol. 19: 150.ArticlePubMedPMCPDF
- Zha L, Li S, Guo J, Hu Y, Pan L, et al. 2025. Global and regional burden of bloodstream infections caused by carbapenem-resistant Gram-negative bacteria in 2019: a systematic analysis from the MICROBE database. Int J Infect Dis. 153: 107769.ArticlePubMed
- Zhang Y, Yang S, Deng Z, Song H, Xie N, et al. 2025. Antifungal agent tavaborole as a potential broad-spectrum serine- and metallo-β-lactamase inhibitor. EBioMedicine. 116: 105754.ArticlePubMedPMC
References
Supplementary Information
References
Citations
- Pioneering strategies for overcoming bacterial drug resistance
Byoung Sik Kim
Journal of Microbiology.2026; 64(3): e2603100. CrossRef
 ePub Link
ePub Link Cite this Article
Cite this Article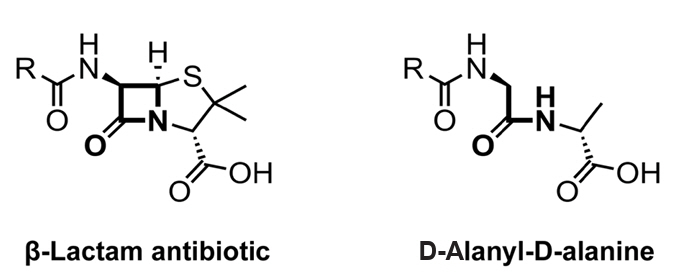
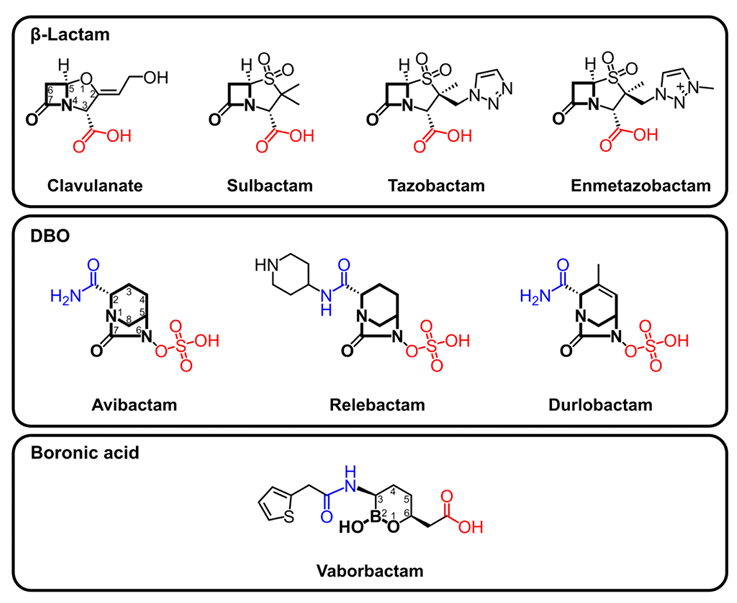
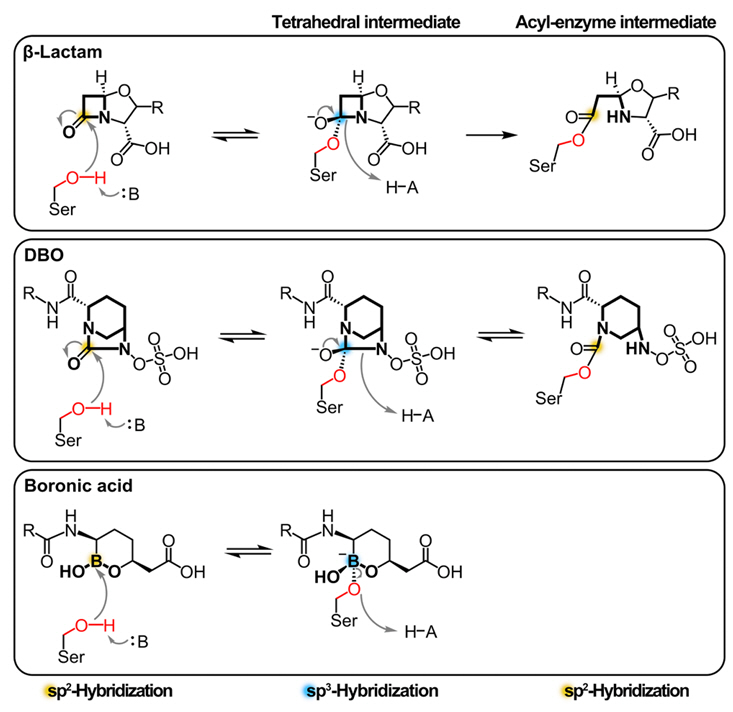
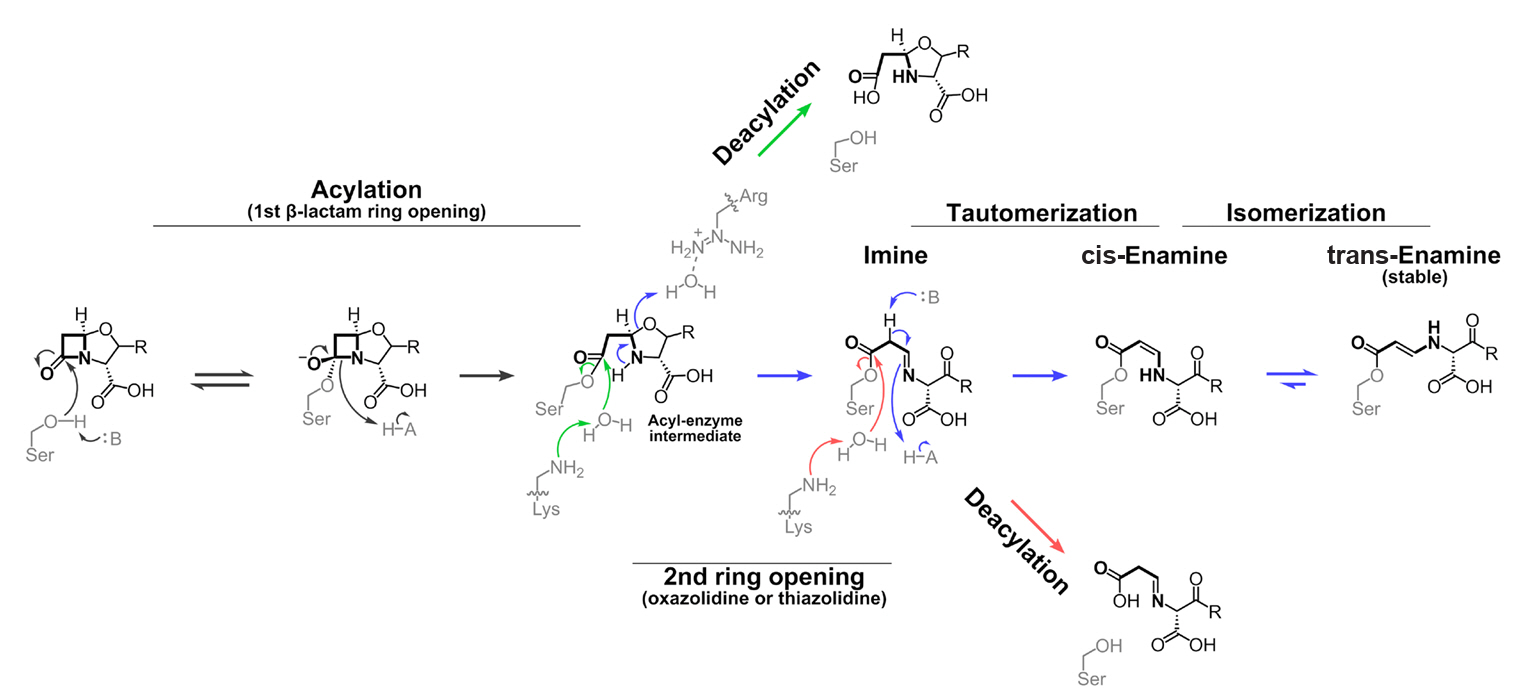
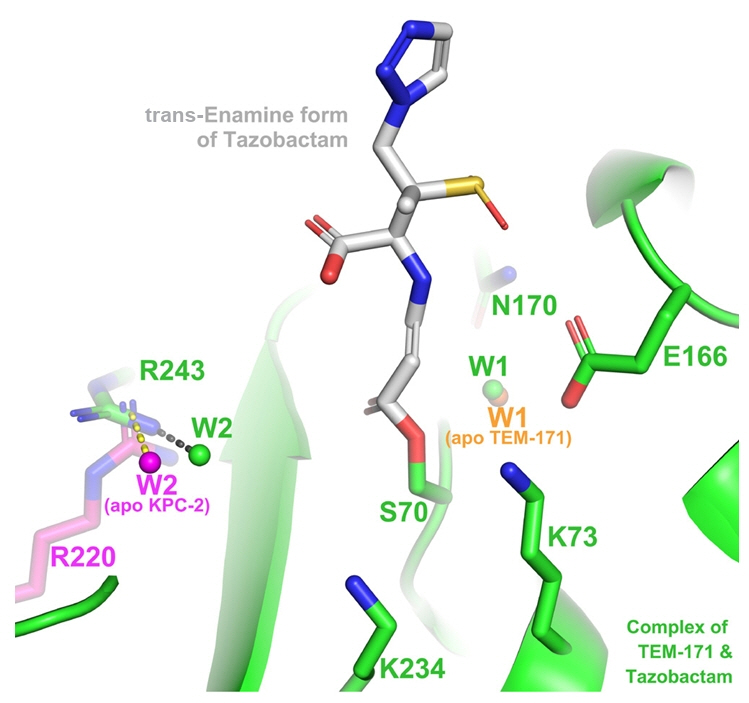
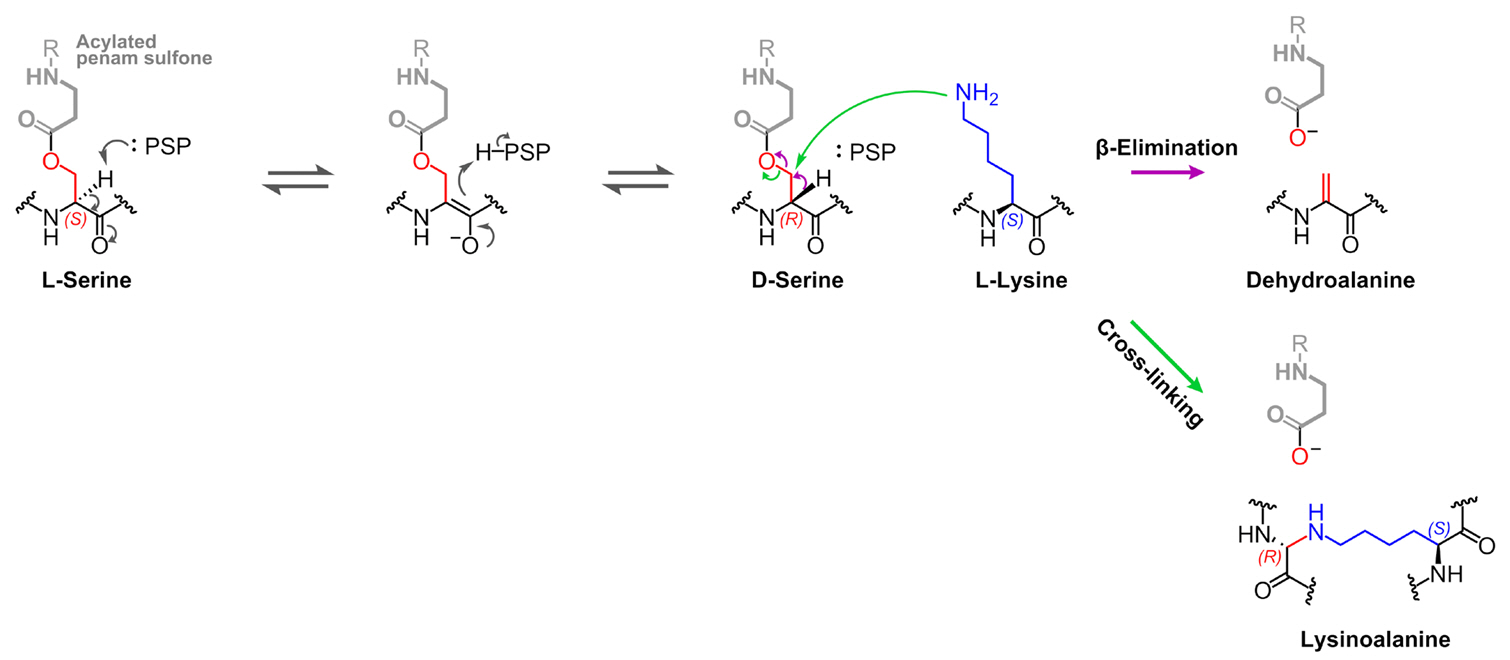
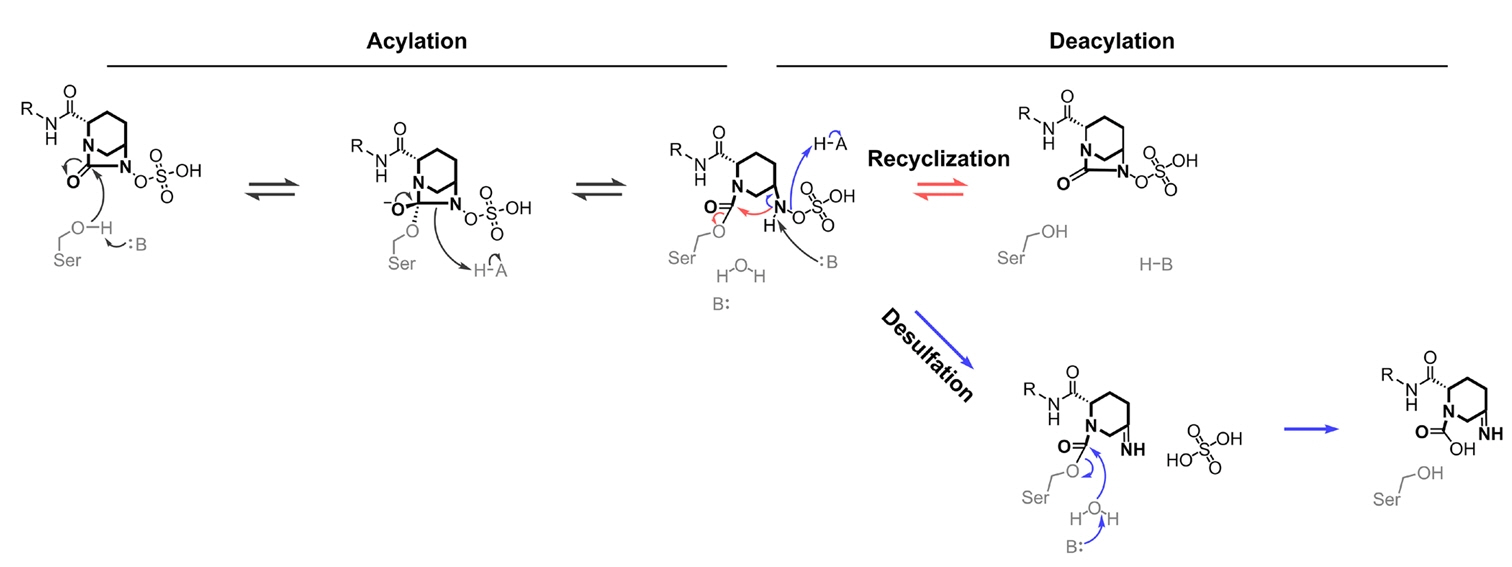
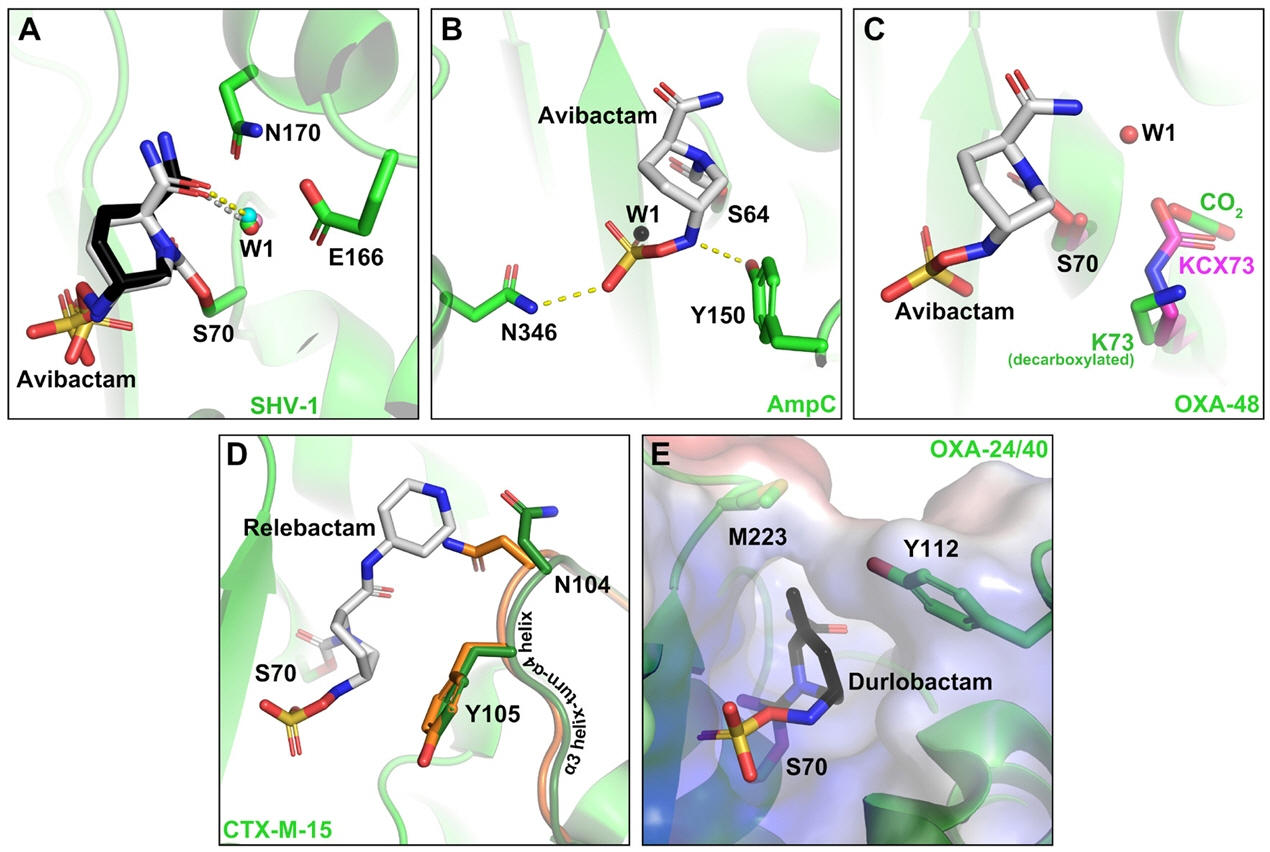
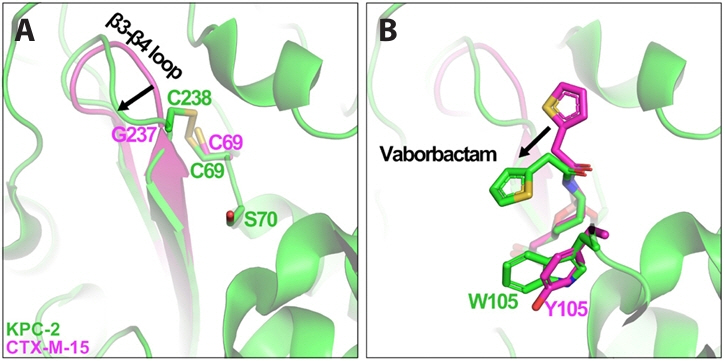
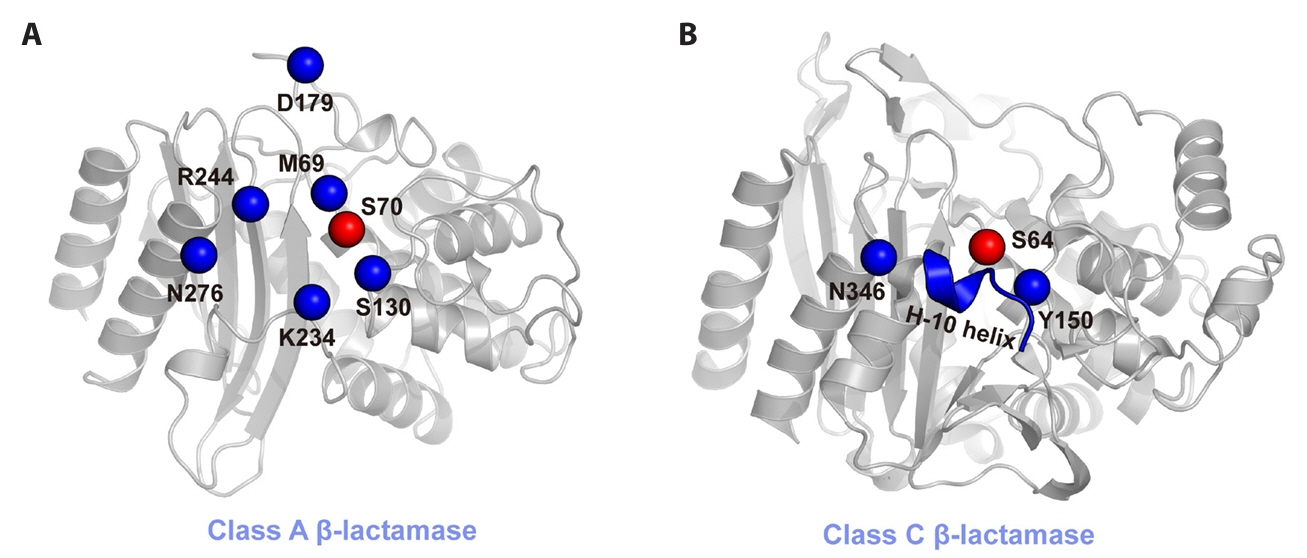
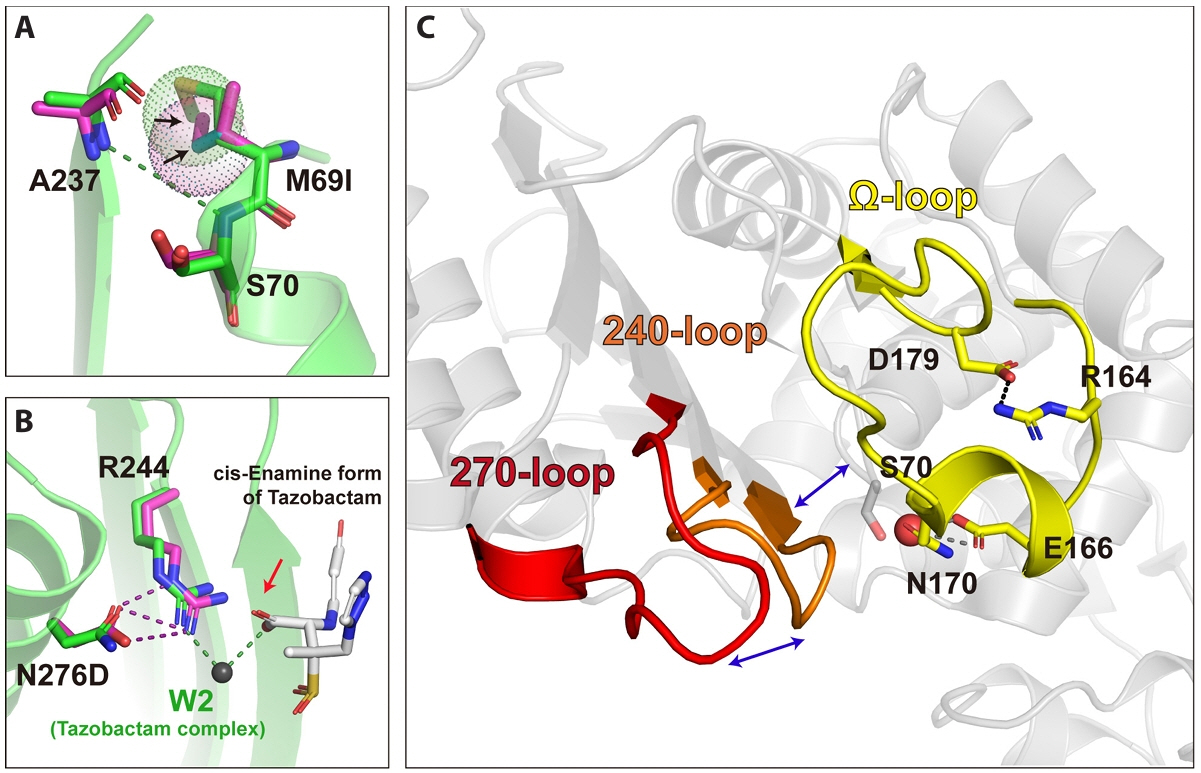















Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
Fig. 7.
Fig. 8.
Fig. 9.
Fig. 10.
Fig. 11.
Fig. 12.
Fig. 13.
Fig. 14.
Fig. 15.
| Core structure | Inhibitor | Partner β-lactam | Formulation (Year of FDA approval) | Inhibition profile of the inhibitor |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Class A | Class C | Class D | Class B | |||||||||
| TEM | SHV | CTX-M | KPC | AmpC | CMY | OXA-23 | OXA-48 | MBL |
||||
| β-Lactam | Clavulanic acid | Amoxicillin | Augmentin (1984) | O | O | O | X | X | X | X | X | X |
| Sulbactam | Ampicillin | Unasyn (1986) | O | △ | △ | X | X | X | X | X | X | |
| Tazobactam | Piperacillin | Zosyn (1993) | O | O | O | X | X | X | X | X | X | |
| Ceftolozane | Zerbaxa (2014) | |||||||||||
| Enmetazobactam | Cefepime | Exblifep (2024) | O | O | O | X | X | X | X | X | X | |
| DBO |
Avibactam | Ceftazidime | Avycaz (2015) | O | O | O | O | O | O | X | O | X |
| Aztreonam | Emblaveo (2025) | |||||||||||
| Relebactam | Imipenem and cilastatin | Recarbrio (2019) | O | O | O | O | O | O | X | X | X | |
| Durlobactam | Sulbactam | Xacduro (2023) | O | O | O | O | O | O | △ | O | X | |
| Boronic acid | Vaborbactam | Meropenem | Vabomere(2017) | O | O | O | O | O | O | X | X | X |
( metallo β-lactamase. diazabicyclooctane. O: potent inhibition; △: moderate inhibition; X: no inhibitory activity.
Table 1.
TOP

