- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- For Contributors
- Policies
- E-Submission

- About
- Browse Articles
-
Special Issues
- Pioneering strategies for overcoming bacterial drug resistance (2026)
- Advancing microbial engineering through synthetic biology (2025)
- Host-associated microbiome (2024)
- Bacterial regulatory mechanisms for the control of complex cellular mechanisms (2023)
- Two years into COVID-19 pandemic: Where are we? (2022)
- Collections
- Policies
- For Contributors
Articles
- Page Path
- HOME > J. Microbiol > Volume 64(6); 2026 > Review
-
Review
From contiguity to accuracy: Validation-centered perspectives on bacterial genome assembly -
Minkyung Kim1, Yong-Joon Cho2,3,*
 , Ok-Sun Kim1,*
, Ok-Sun Kim1,* -
Journal of Microbiology 2026;64(6):e2604004.
DOI: https://doi.org/10.71150/jm.2604004
Published online: June 19, 2026
1Division of Life Sciences, Korea Polar Research Institute, Incheon 21990, Republic of Korea
2Department of Molecular Bioscience, Kangwon National University, Chuncheon 24341, Republic of Korea
3Multidimensional Genomics Research Center, Kangwon National University, Chuncheon 24341, Republic of Korea
- *Correspondence Yong-Joon Cho yongjoon@kangwon.ac.kr Ok-Sun Kim oskim@kopri.re.kr
© The Author(s), under exclusive licence to Microbiological Society of Korea 2026
This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0) (https://creativecommons.org/licenses/by/4.0/) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,484 Views
- 32 Download
- ABSTRACT
- Introduction
- Structural Ambiguity Arising from Complex Genome Architecture
- Limitations of Long-Read Sequencing Accuracy
- Challenges and Limitations of Genome Assembly Algorithms
- Polishing and Validation Approaches for Genome Assembly
- A Practical Framework for High-Confidence Complete Genomes
- Future Perspectives
- Notes
- Supplementary Information
- References
ABSTRACT
- Recent advances in sequencing technologies, particularly long-read platforms, have substantially improved contiguity of bacterial genome assemblies and enabled the routine generation of near-complete or circular genomes. However, achieving a contiguous assembly does not necessarily guarantee accuracy. Assembly errors, including structural misassemblies, collapsed repeats, incorrect circularization, plasmid reconstruction errors, and nucleotide-level inaccuracies, remain prevalent and may lead to misleading biological interpretations if not properly identified. In this review, we provide a comprehensive overview of bacterial genome assembly from a validation-centered perspective and examine the underlying causes of draft genome formation and assembly uncertainty, highlighting the roles of repetitive genomic structures, platform-specific error profiles, and algorithmic limitations. We further emphasize that the central challenge in contemporary bacterial genomics is no longer simply to maximize assembly contiguity, but to determine whether apparently complete genomes are truly correct and sufficiently reliable for their intended downstream applications. We propose a practical decision-making framework that links sequencing strategy, assembly workflow, polishing, and validation rigor, and introduce a tiered confidence classification to guide the interpretation of genome assembly reliability. As bacterial genome sequencing becomes increasingly routine and large-scale, future efforts should prioritize accuracy, reproducibility, transparent reporting, and evidence-supported validation over completeness alone.
Introduction
Structural Ambiguity Arising from Complex Genome Architecture
Limitations of Long-Read Sequencing Accuracy
Challenges and Limitations of Genome Assembly Algorithms
Polishing and Validation Approaches for Genome Assembly
A Practical Framework for High-Confidence Complete Genomes
Future Perspectives
Acknowledgments
This work was supported by the Korea Polar Research Institute (KOPRI) [PE26100] and by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2026-25478655 and RS-2026-25491179). This work was also supported by Global-Learning & Academic research institution for Master, PhD students, and Postdocs (G-LAMP) Program (RS-2023-00301850).
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
Table S1.
| Tool | Input | Core engine | Note | Reference |
|---|---|---|---|---|
| ARACHNE | Short (Sanger) | OLC | Efficient scaling; primarily suited for large eukaryotic genomes | Batzoglou et al. (2002) |
| PCAP | Short (Sanger) | OLC (Sanger-era) | Parallel Sanger-era assembler; large-genome oriented | Huang et al. (2003) |
| Newbler | Short (454) | OLC | Widely used in early bacterial genome assembly | Margulies et al. (2005) |
| Mira | Short | OLC | Supports mapping-based assembly and polishing | Chevreux (2005) |
| PHRAP | Short (Sanger) | OLC (Sanger-era) | Sanger-era shotgun assembler | de la Bastide and McCombie (2007) |
| EULER-SR | Short (454) | DBG | A-Bruijn–inspired DBG | Chaisson and Pevzner (2008) |
| Velvet | Short | DBG | Early short-read DBG assembler | Zerbino and Birney (2008) |
| ALLPATHS-LG | Short | DBG | Optional long-read integration for gap filling | MacCallum et al. (2009) |
| IDBA | Short | Iterative DBG | Handles uneven coverage | Peng et al. (2010) |
| SOAPdenovo | Short | DBG | Scaffold-oriented; widely used for large genomes | Li et al. (2010) |
| SOAPdenovo2 | Short | DBG | Improved memory efficiency and accuracy | Luo et al. (2012) |
| Minia version 3 | Short | Compacted DBG | Unitig-based; evolved from Bloom filter–based approach | Salikhov et al. (2013) |
| MEGAHIT* | Short | Succinct DBG | Optimized for metagenome assembly | Li et al. (2015) |
| SPAdes* | Short | DBG | Multisized and paired-end integration | Prjibelski et al. (2020) |
| HybridSPAdes* | Hybrid | DBG | Multisized, paired-end, and long-read integration | Antipov et al. (2016b) |
| FALCON | Long | String graph | Diploid-aware; optimized for complex eukaryotic genomes | Chin et al. (2016) |
| Miniasm | Long | OLC | Consensus-free; requires polishing | Li (2016) |
| HINGE | Long | OLC (repeat-aware) | Improves repeat resolution using hinge-based graph construction | Kamath et al. (2017) |
| Canu* | Long | OLC | Designed for noisy long reads | Koren et al. (2017) |
| Flye* | Long | Repeat graph | Robust to complex repeats | Kolmogorov et al. (2019) |
| HiCanu* | Long (HiFi) | OLC | Improved accuracy and repeat resolution | Nurk et al. (2020) |
| wtdbg2* | Long | Fuzzy Bruijn graph | Fast and memory-efficient; designed for noisy long reads | Ruan and Li (2020) |
| Shasta* | Long (ONT) | Marker graph | Fast and memory-efficient | Shafin et al. (2020) |
| Raven* | Long | OLC | Optimized for long uncorrected reads; fast and memory-efficient | Vaser and Šikić (2021) |
| Hifiasm* | Long (HiFi) | String graph | High accuracy and repeat resolution | Cheng et al. (2021) |
| NECAT* | Long (ONT) | OLC | Efficient assembly of noisy long reads | Chen et al. (2021a) |
| SmartDenovo | Long | OLC | No error correction | Liu et al. (2021) |
| NextDenovo* | Long | OLC | Improved accuracy | Hu et al. (2024b) |
| Tool | Category | Input | Core method | Key function | Reference |
|---|---|---|---|---|---|
| Pilon | Short-read polishing | Illumina reads | Read mapping | Error correction of SNPs and small indels | Walker et al. (2014) |
| Polypolish | Short-read polishing | Illumina reads | Multi-mapping | Repeat-aware error correction | Wick and Holt (2022) |
| Pypolca | Short-read polishing | Illumina reads | Read mapping | Error correction with threshold-based variant filtering | Bouras et al. (2024b) |
| Racon | Long-read polishing | Long reads | Read mapping | Consensus-based error correction | Vaser et al. (2017) |
| NeuralPolish | Long-read polishing | Long reads | Deep learning | Improved base accuracy using neural networks | Huang et al. (2021) |
| DeepPolisher | Long-read polishing | Long reads | Deep learning | Deep learning–based error correction | Mastoras et al. (2025) |
| Medaka | Long-read polishing | ONT reads | Deep learning | Signal-aware error correction | Medaka (2018) |
| NextPolish | Hybrid polishing | Short + long reads | Iterative polishing | Multi-platform error correction | Hu et al. (2020) |
| Tool | Category | Input | Core method | Key function | Reference |
|---|---|---|---|---|---|
| REAPR | Read-based | Short reads + assembly | Read mapping | Error detection | Hunt et al. (2013) |
| Inspector | Read-based | Long reads | Read mapping | Structural and local error detection with correction | Chen et al. (2021b) |
| QUAST | Reference-based | Assembly + reference | Whole-genome alignment | Assembly quality assessment and misassembly detection | Gurevich et al. (2013) |
| Assemblytics | Reference-based | Assembly + reference | Whole-genome alignment | Structural variation detection | Nattestad and Schatz (2016) |
| CheckM | Completeness | Assembly | Lineage-specific marker genes | Completeness + contamination | Parks et al. (2015) |
| BUSCO | Completeness | Assembly | Marker genes (Single-copy orthologs) | Completeness assessment | Seppey et al. (2019) |
| KAT | k-mer based | Reads + assembly | k-mer comparison | Coverage bias, duplication detection | Mapleson et al. (2017) |
| Merqury | k-mer based | Reads + assembly | k-mer spectrum | Accuracy (QV) + completeness | Rhie et al. (2020) |
| Bandage | Graph-based | Assembly graph | Visualization | Graph inspection | Wick et al. (2015) |
| gfatools | Graph-based | GFA | Graph parsing | Structural analysis | Pani et al. (2024) |
| Circlator | Structural | Assembly | Overlap detection | Circularization validation | Hunt et al. (2015) |
| MOB-suite | Plasmid | Assembly | Database + typing | Plasmid reconstruction | Robertson and Nash (2018) |
- Acuña-Amador L, Primot A, Cadieu E, Roulet A, Barloy-Hubler F. 2018. Genomic repeats, misassembly and reannotation: A case study with long-read resequencing of Porphyromonas gingivalis reference strains. BMC Genomics. 19: 54.ArticlePubMedPMC
- Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, et al. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21: 30.ArticlePubMedPMCPDF
- Antipov D, Hartwick N, Shen M, Raiko M, Ladipus A, et al. 2016a. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics. 32: 3380–3387. ArticlePDF
- Antipov D, Korobeynikov A, McLean JS, Pevzner PA. 2016b. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics. 32: 1009–1015. Article
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. 2012. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19: 455–477. ArticlePubMedPMCLink
- Batzoglou S, Jaffe DB, Stanley K, Butler J, Gnerre S, et al. 2002. ARACHNE: A whole-genome shotgun assembler. Genome Res. 12: 177–189. ArticlePubMedPMC
- Bentley SD, Maiwald M, Murphy LD, Pallen MJ, Yeats CA, et al. 2003. Sequencing and analysis of the genome of the Whipple's disease bacterium Tropheryma whipplei. Lancet. 361: 637–644. ArticlePubMed
- Bouras G, Houtak G, Wick RR, Mallawaarachchi V, Roach MJ, et al. 2024a. Hybracter: Enabling scalable, automated, complete and accurate bacterial genome assemblies. Microb Genom. 10: 001244.Article
- Bouras G, Judd LM, Edwards RA, Vreugde S, Stinear TP, et al. 2024b. How low can you go? Short-read polishing of Oxford Nanopore bacterial genome assemblies. Microb Genom. 10: 001254.Article
- Branton D, Deamer DW. 2019. Nanopore sequencing: An introduction. World Scientific PublishingLink
- Bzikadze AV, Mikheenko A, Pevzner PA. 2022. Fast and accurate mapping of long reads to complete genome assemblies with VerityMap. Genome Res. 32: 2107–2118. ArticlePubMedPMC
- Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, et al. 2014. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 58: 3895–3903. ArticlePubMedPMCLink
- Chaisson MJ, Pevzner PA. 2008. Short read fragment assembly of bacterial genomes. Genome Res. 18: 324–330. ArticlePubMed
- Chen Y, Nie F, Xie SQ, Zheng YF, Dai Q, et al. 2021a. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat Commun. 12: 60.ArticlePDF
- Chen Y, Zhang Y, Wang AY, Gao M, Chong Z. 2021b. Accurate long-read de novo assembly evaluation with Inspector. Genome Biol. 22: 312.ArticlePDF
- Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18: 170–175. ArticlePubMedPMCPDF
- Chevreux B. 2005. Ph.D. thesis. MIRA: An automated genome and EST assembler. The Ruprecht-Karls-University, Heidelberg, Germany.PDF
- Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, et al. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 13: 1050–1054. ArticlePubMedPMCPDF
- Chiou CS, Chen BH, Wang YW, Kuo NT, Chang CH, et al. 2023. Correcting modification-mediated errors in nanopore sequencing by nucleotide demodification and reference-based correction. Commun Biol. 6: 1215.ArticlePubMedPMCPDF
- Colombini L, Santoro F, Tirziu M, Cuppone AM, Pozzi G, et al. 2025. A 69.9-kb long inverted repeat increases genome instability in a strain of Lactobacillus crispatus. NAR Genom Bioinform. 7: lqaf085.ArticlePubMedPMCPDF
- de la Bastide M, McCombie WR. 2007. Assembling genomic DNA sequences with PHRAP. Curr Protoc Bioinformatics. 17: 11.4.1–11.4.15. Article
- Delahaye C, Nicolas J. 2021. Sequencing DNA with nanopores: Troubles and biases. PLoS One. 16: e0257521. ArticlePubMedPMC
- Di Genova A, Buena-Atienza E, Ossowski S, Sagot MF. 2021. Efficient hybrid de novo assembly of human genomes with WENGAN. Nat Biotechnol. 39: 422–430. ArticlePubMedPDF
- Eiglmeier K, Parkhill J, Honoré N, Garnier T, Tekaia F, et al. 2001. The decaying genome of Mycobacterium leprae. Lepr Rev. 72: 387–398. ArticlePubMed
- El Kafsi H, Loux V, Mariadassou M, Blin C, Chiapello H, et al. 2017. Unprecedented large inverted repeats at the replication terminus of circular bacterial chromosomes suggest a novel mode of chromosome rescue. Sci Rep. 7: 44331.ArticlePubMedPMC
- Espinosa E, Bautista R, Fernandez I, Larrosa R, Zapata EL, et al. 2023. Comparing assembly strategies for third-generation sequencing technologies across different genomes. Genomics. 115: 110700.ArticlePubMed
- Firtina C, Kim JS, Alser M, Senol Cali D, Cicek AE, et al. 2020. Apollo: A sequencing-technology-independent, scalable and accurate assembly polishing algorithm. Bioinformatics. 36: 3669–3679. ArticlePubMedPDF
- Gao S, Tran Q, Phan V. 2019. Understand effective coverage by mapped reads using genome repeat complexity. Proceedings of 11th International Conference on Bioinformatics and Computational Biology, BiCOB. 65–73 Available from https://digitalcommons.memphis.edu/facpubs/3302. Article
- Glaser P, Kunst F, Arnaud M, Coudart MP, Gonzales W, et al. 1993. Bacillus subtilis genome project: Cloning and sequencing of the 97 kb region from 325 degrees to 333 degrees. Mol Microbiol. 10: 371–384. Article
- Gunasekera S, Abraham S, Stegger M, Pang S, Wang P, et al. 2021. Evaluating coverage bias in next-generation sequencing of Escherichia coli. PLoS One. 16: e0253440. ArticlePubMedPMC
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 29: 1072–1075. ArticlePubMedPMCPDF
- Hon T, Mars K, Young G, Tsai YC, Karalius JW, et al. 2020. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data. 7: 399.ArticlePubMedPMCPDF
- Hu J, Fan J, Sun Z, Liu S. 2020. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36: 2253–2255. ArticlePubMedPDF
- Hu J, Wang Z, Liang F, Liu SL, Ye K, et al. 2024a. NextPolish2: A repeat-aware polishing tool for genomes assembled using HiFi long reads. Genomics Proteomics Bioinformatics. 22: qzad009.ArticlePDF
- Hu J, Wang Z, Sun Z, Hu B, Ayoola AO, et al. 2024b. NextDenovo: An efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25: 107.ArticlePDF
- Huang N, Nie F, Ni P, Luo F, Gao X, et al. 2021. NeuralPolish: A novel nanopore polishing method based on alignment matrix construction and orthogonal Bi-GRU networks. Bioinformatics. 37: 3120–3127. ArticlePubMedPDF
- Huang X, Wang J, Aluru S, Yang SP, Hillier L. 2003. PCAP: A whole-genome assembly program. Genome Res. 13: 2164–2170. ArticlePubMedPMC
- Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, et al. 2013. REAPR: A universal tool for genome assembly evaluation. Genome Biol. 14: R47.ArticlePubMedPMCPDF
- Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, et al. 2015. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16: 294.ArticlePubMedPMCPDF
- Jain C. 2023. Coverage-preserving sparsification of overlap graphs for long-read assembly. Bioinformatics. 39: btad124.ArticlePubMedPMC
- Johnson J, Soehnlen M, Blankenship HM. 2023. Long read genome assemblers struggle with small plasmids. Microb Genom. 9: mgen001024.ArticlePubMedPMC
- Kamath GM, Shomorony I, Xia F, Courtade TA, Tse DN. 2017. HINGE: Long-read assembly achieves optimal repeat resolution. Genome Res. 27: 747–756. ArticlePubMedPMC
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 37: 540–546. ArticlePubMedPDF
- Koren S, Harhay GP, Smith TP, Bono JL, Harhay DM, et al. 2013. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 14: R101.ArticlePubMedPMCPDF
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, et al. 2017. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27: 722–736. ArticlePubMedPMC
- Lee JY, Kong M, Oh J, Lim J, Chung SH, et al. 2021. Comparative evaluation of nanopore polishing tools for microbial genome assembly and polishing strategies for downstream analysis. Sci Rep. 11: 20740.ArticlePubMedPMCPDF
- Lerminiaux N, Fakharuddin K, Mulvey MR, Mataseje L. 2024. Do we still need Illumina sequencing data? Evaluating Oxford Nanopore Technologies R10.4.1 flow cells and the Rapid v14 library prep kit for Gram negative bacteria whole genome assemblies. Can J Microbiol. 70: 178–189. ArticlePubMed
- Li H. 2016. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics. 32: 2103–2110. ArticlePubMedPMCPDF
- Li D, Liu CM, Luo R, Sadakane K, Lam TW. 2015. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31: 1674–1676. ArticlePubMedPDF
- Li K, Xu P, Wang J, Yi X, Jiao Y. 2023. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat Commun. 14: 6556.ArticlePubMedPMCPDF
- Li R, Zhu H, Ruan J, Qian W, Fang X, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20: 265–272. ArticlePubMed
- Liu H, Wu S, Li A, Ruan J. 2021. SMARTdenovo: A de novo assembler using long noisy reads. GigaByte. 2021: gigabyte15.ArticlePubMedPMC
- Loman NJ, Pallen MJ. 2015. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 13: 787–794. ArticlePubMedPDF
- Luan T, Commichaux S, Hoffmann M, Jayeola V, Jang JH, et al. 2024. Benchmarking short and long read polishing tools for nanopore assemblies: Achieving near-perfect genomes for outbreak isolates. BMC Genomics. 25: 679.ArticlePubMedPMCPDF
- Luo R, Liu B, Xie Y, Li Z, Huang W, et al. 2012. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience. 1: 18.ArticlePubMedPMC
- MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, et al. 2009. ALLPATHS 2: Small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 10: R103.ArticlePubMedPMCPDF
- Mäkinen V, Salmela L, Ylinen J. 2012. Normalized N50 assembly metric using gap-restricted co-linear chaining. BMC Bioinformatics. 13: 255.ArticlePubMedPMC
- Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J, Clavijo BJ. 2017. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 33: 574–576. ArticlePubMedPMCPDF
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 437: 376–380. ArticlePubMedPMC
- Marijon P, Chikhi R, Varré JS. 2019. Graph analysis of fragmented long-read bacterial genome assemblies. Bioinformatics. 35: 4239–4246. ArticlePubMedPDF
- Mastoras M, Asri M, Brambrink L, Hebbar P, Kolesnikov A, et al. 2025. Highly accurate assembly polishing with DeepPolisher. Genome Res. 35: 1595–1608. ArticlePubMedPMC
- Medaka. 2018. Sequence correction provided by ONT Research. Available from https://github.com/nanoporetech/medaka (accessed April 2026)Link
- Merda D, Vila-Nova M, Bonis M, Boutigny AL, Brauge T, et al. 2024. Unraveling the impact of genome assembly on bacterial typing: A one health perspective. BMC Genomics. 25: 1059.ArticlePubMedPMCPDF
- Metzker M. 2010. Sequencing technologies — the next generation. Nat Rev Genet. 11: 31–46. ArticlePubMedPDF
- Miller JR, Koren S, Sutton G. 2010. Assembly algorithms for next-generation sequencing data. Genomics. 95: 315–327. ArticlePubMedPMC
- Molina-Mora JA, Campos-Sánchez R, Rodríguez C, Shi L, García F. 2020. High quality 3C de novo assembly and annotation of a multidrug resistant ST-111 Pseudomonas aeruginosa genome: Benchmark of hybrid and non-hybrid assemblers. Sci Rep. 10: 1392.ArticlePubMedPMCPDF
- Myers EW. 2005. The fragment assembly string graph. Bioinformatics. 21: ii79–ii85. ArticlePubMedPDF
- Nagarajan N, Pop M. 2013. Sequence assembly demystified. Nat Rev Genet. 14: 157–167. ArticlePubMedPDF
- Nattestad M, Schatz MC. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics. 32: 3021–3023. ArticlePubMedPMCPDF
- NCBI. Genome datasets. 2026. Available from https://www.ncbi.nlm.nih.gov/datasets/genome/ (accessed April 2026)Link
- Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, et al. 2020. HiCanu: Accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30: 1291–1305. ArticlePubMedPMC
- Pani S, Dabbaghie F, Marschall T, Söylev A. 2024. A toolkit for analyzing and manipulating pangenome alignments. bioRxiv. doi: https://doi.org/10.1101/2024.12.10.627813. Article
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25: 1043–1055. ArticlePubMedPMC
- Peng Y, Leung HCM, Yiu SM, Chin FYL. 2010. IDBA—A practical iterative de Bruijn graph de novo assembler. In Berger B. (ed.), Research in computational molecular biology, vol. 6044, pp. 426–440. Springer. Article
- Peng K, Li C, Wang Q, Xin X, Wang Z, et al. 2025. The applications and advantages of nanopore sequencing in bacterial antimicrobial resistance surveillance and research. NPJ Antimicrob Resist. 3: 87.ArticlePubMedPMCPDF
- Phillippy AM, Schatz MC, Pop M. 2008. Genome assembly forensics: Finding the elusive mis-assembly. Genome Biol. 9: R55.ArticlePubMedPMCPDF
- Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. 2020. Using SPAdes de novo assembler. Curr Protoc Bioinformatics. 70: e102. ArticlePubMedLink
- Reuter JA, Spacek DV, Snyder MP. 2015. High-throughput sequencing technologies. Mol Cell. 58: 586–597. ArticlePubMedPMC
- Rhie A, Walenz BP, Koren S, Phillippy AM. 2020. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21: 245.ArticlePubMedPMCPDF
- Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genom Proteom Bioinform. 13: 278–289. ArticlePubMedPMCPDF
- Robertson J, Nash JHE. 2018. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom. 4: e000206. ArticlePubMedPMC
- Rojas-Miranda H, Madrigal-Ly V, Molina-Mora JA. 2025. Benchmarking genome assemblers for four bacterial models based on contiguity, correctness, and completeness. Sci Rep. 15: 42858.ArticlePubMedPMCPDF
- Ruan J, Li H. 2020. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 17: 155–158. ArticlePubMedPDF
- Salikhov K, Sacomoto G, Kucherov G. 2013. Using cascading Bloom filters to improve the memory usage for de Bruijn graphs. Algorithms Mol Biol. 9: 2.Article
- Seppey M, Manni M, Zdobnov EM. 2019. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol Biol. 1962: 227–245. ArticlePubMed
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, et al. 2020. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 38: 1044–1053. ArticlePubMedPDF
- Shintani M, Sanchez ZK, Kimbara K. 2015. Genomics of microbial plasmids: Classification and identification based on replication and transfer systems and host taxonomy. Front Microbiol. 6: 242.ArticlePubMedPMC
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31: 3210–3212. ArticlePubMedPDF
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, et al. 2009. ABySS: A parallel assembler for short read sequence data. Genome Res. 19: 1117–1123. ArticlePubMedPMC
- Sousa TJ, Parise D, Profeta R, Parise MTD, Gomide ACP, et al. 2019. Re-sequencing and optical mapping reveals misassemblies and real inversions on Corynebacterium pseudotuberculosis genomes. Sci Rep. 9: 16387.ArticlePubMedPMCPDF
- Thrash A, Hoffmann F, Perkins A. 2020. Toward a more holistic method of genome assembly assessment. BMC Bioinformatics. 21: 249.ArticlePubMedPMCPDF
- Tizabi D, Bachvaroff T, Hill RT. 2022. Comparative analysis of assembly algorithms to optimize biosynthetic gene cluster identification in novel marine actinomycete genomes. Front Mar Sci. 9: 914197.Article
- Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. 2010. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 38: e159. ArticlePubMedPMC
- Treangen TJ, Abraham AL, Touchon M, Rocha EP. 2009. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol Rev. 33: 539–571. ArticlePubMed
- Treangen TJ, Salzberg SL. 2011. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat Rev Genet. 13: 36–46. ArticlePubMedPMCPDF
- Trisakul K, Hinwan Y, Eisiri J, Salao K, Chaiprasert A, et al. 2024. Comparisons of genome assembly tools for characterization of Mycobacterium tuberculosis genomes using hybrid sequencing technologies. PeerJ. 12: e17964. ArticlePubMedPMCPDF
- Vaser R, Šikić M. 2021. Time- and memory-efficient genome assembly with Raven. Nat Comput Sci. 1: 332–336. ArticlePubMedPDF
- Vaser R, Sović I, Nagarajan N, Šikić M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27: 737–746. ArticlePubMedPMC
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, et al. 2014. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9: e112963. ArticlePubMedPMC
- Waters EV, Cameron SK, Langridge GC, Preston A. 2025. Bacterial genome structural variation: Prevalence, mechanisms, and consequences. Trends Microbiol. 33: 875–886. ArticlePubMed
- Watson M, Warr A. 2019. Errors in long-read assemblies can critically affect protein prediction. Nat Biotechnol. 37: 124–126. ArticlePubMedPDF
- Wenger AM, Peluso P, Rowell WJ, Chang PC, Hall RJ, et al. 2019. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 37: 1155–1162. ArticlePubMedPMCPDF
- Wick RR, Holt KE. 2021. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Res. 8: 2138.ArticleLink
- Wick RR, Holt KE. 2022. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLoS Comput Biol. 18: e1009802. ArticlePubMedPMC
- Wick RR, Howden BP, Stinear TP. 2025. Autocycler: Long-read consensus assembly for bacterial genomes. Bioinformatics. 41: btaf474.ArticlePubMedPMC
- Wick RR, Judd LM, Cerdeira LT, Hawkey J, Méric G, et al. 2021a. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 22: 266.ArticlePDF
- Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 13: e1005595. ArticlePubMedPMC
- Wick RR, Judd LM, Holt KE. 2023. Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput Biol. 19: e1010905. ArticlePubMedPMC
- Wick RR, Judd LM, Wyres KL, Holt KE. 2021b. Recovery of small plasmid sequences via Oxford Nanopore sequencing. Microb Genom. 7: 000631.Article
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics. 31: 3350–3352. ArticlePubMedPMCPDF
- Zerbino DR, Birney E. 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18: 821–829. ArticlePubMedPMC
References
Supplementary Information
References
Citations
 ePub Link
ePub Link Cite this Article
Cite this Article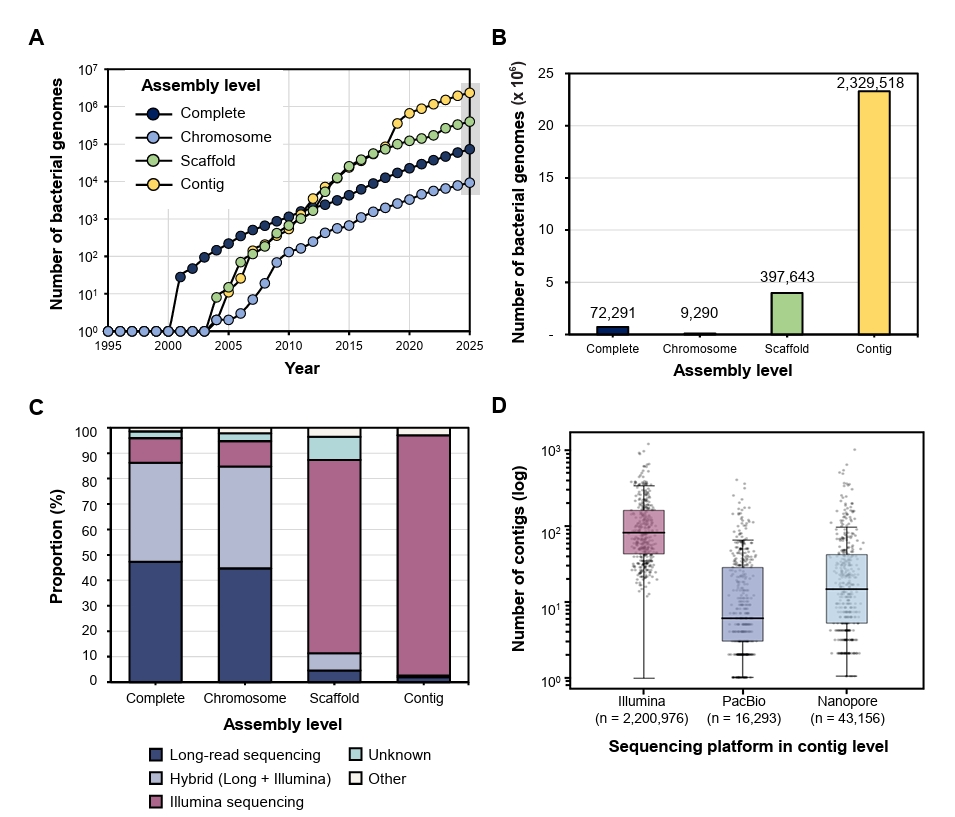
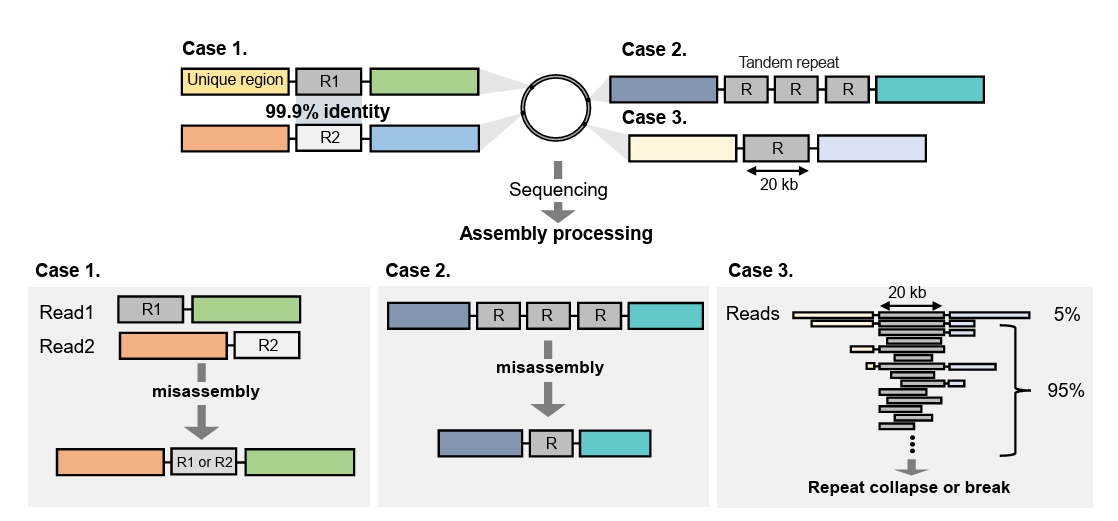
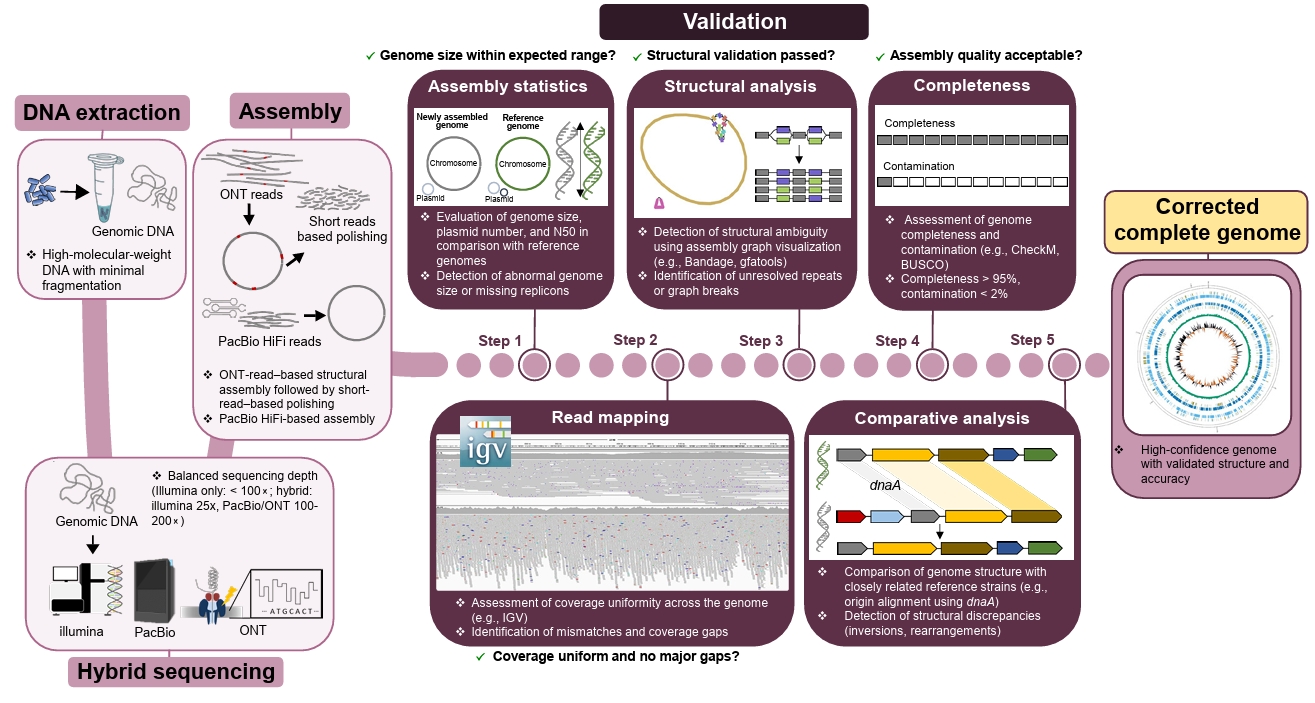



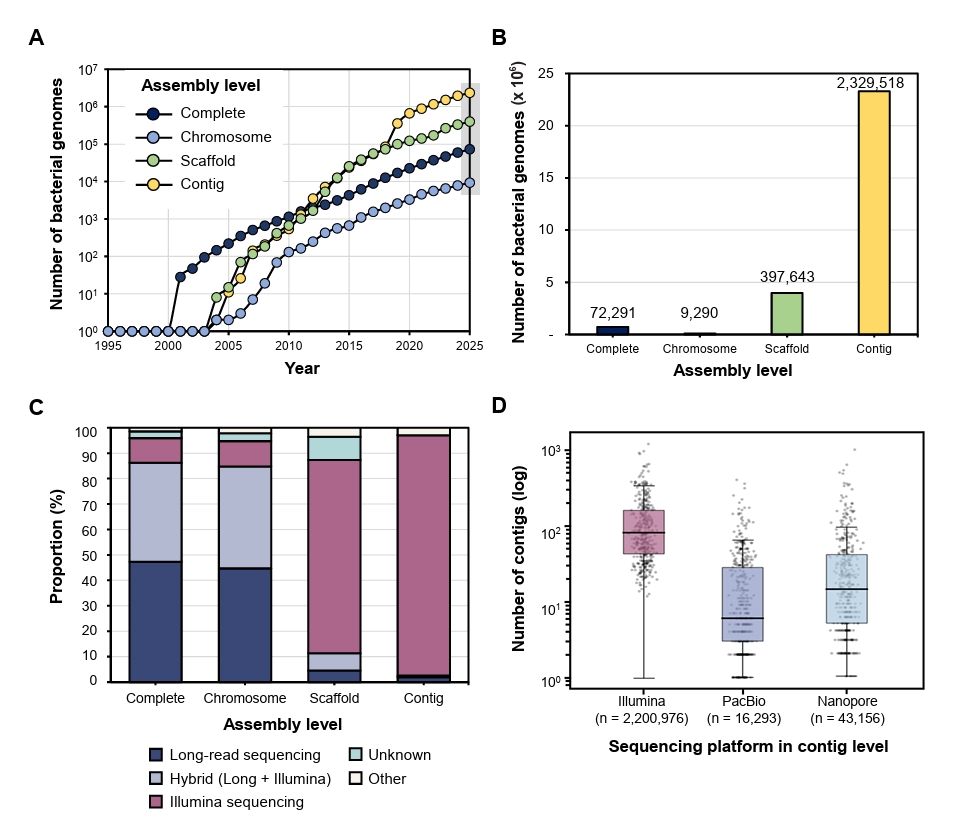
Fig. 1.
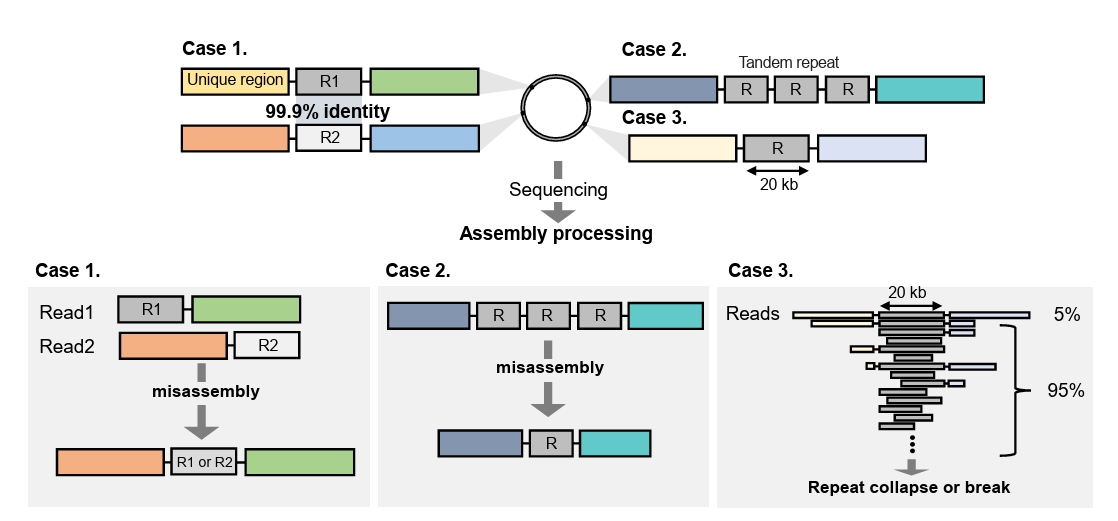
Fig. 2.
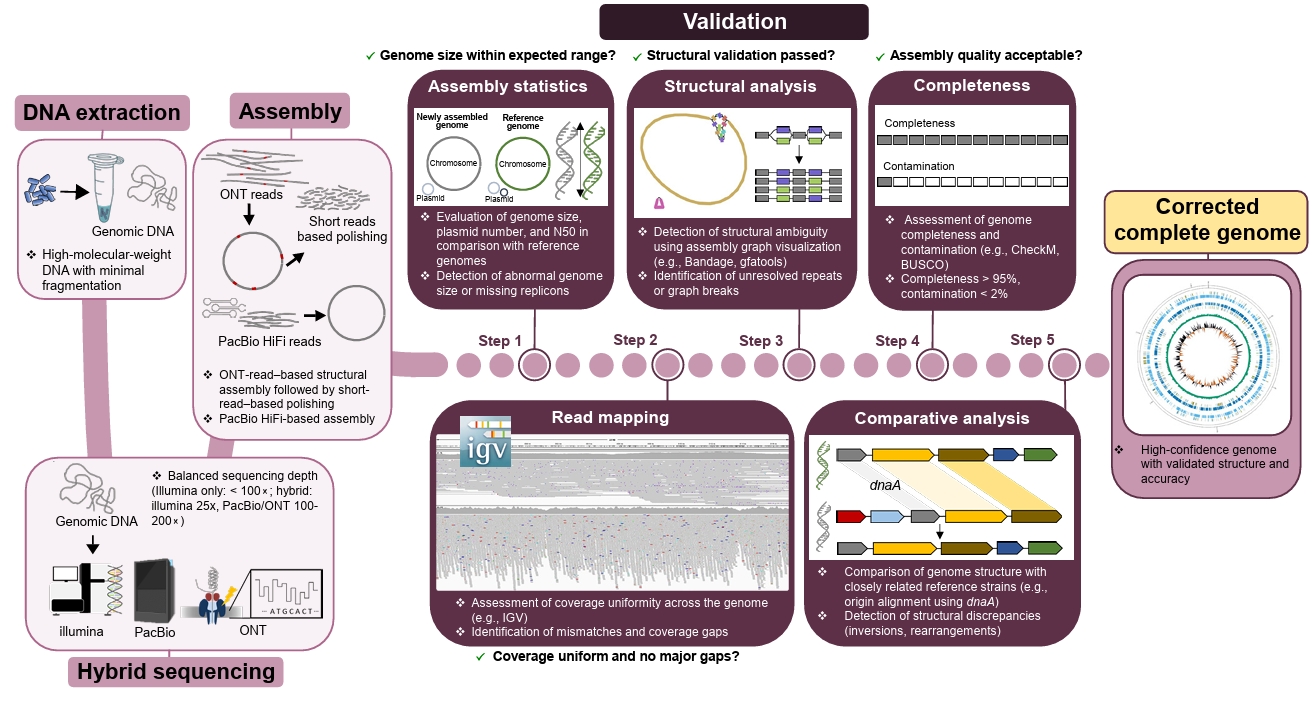
Fig. 3.
| Tool | Input | Core engine | Note | Reference |
|---|---|---|---|---|
| ARACHNE | Short (Sanger) | OLC | Efficient scaling; primarily suited for large eukaryotic genomes | |
| PCAP | Short (Sanger) | OLC (Sanger-era) | Parallel Sanger-era assembler; large-genome oriented | |
| Newbler | Short (454) | OLC | Widely used in early bacterial genome assembly | |
| Mira | Short | OLC | Supports mapping-based assembly and polishing | |
| PHRAP | Short (Sanger) | OLC (Sanger-era) | Sanger-era shotgun assembler | |
| EULER-SR | Short (454) | DBG | A-Bruijn–inspired DBG | |
| Velvet | Short | DBG | Early short-read DBG assembler | |
| ALLPATHS-LG | Short | DBG | Optional long-read integration for gap filling | |
| IDBA | Short | Iterative DBG | Handles uneven coverage | |
| SOAPdenovo | Short | DBG | Scaffold-oriented; widely used for large genomes | |
| SOAPdenovo2 | Short | DBG | Improved memory efficiency and accuracy | |
| Minia version 3 | Short | Compacted DBG | Unitig-based; evolved from Bloom filter–based approach | |
| MEGAHIT |
Short | Succinct DBG | Optimized for metagenome assembly | |
| SPAdes |
Short | DBG | Multisized and paired-end integration | |
| HybridSPAdes |
Hybrid | DBG | Multisized, paired-end, and long-read integration | |
| FALCON | Long | String graph | Diploid-aware; optimized for complex eukaryotic genomes | |
| Miniasm | Long | OLC | Consensus-free; requires polishing | |
| HINGE | Long | OLC (repeat-aware) | Improves repeat resolution using hinge-based graph construction | |
| Canu |
Long | OLC | Designed for noisy long reads | |
| Flye |
Long | Repeat graph | Robust to complex repeats | |
| HiCanu |
Long (HiFi) | OLC | Improved accuracy and repeat resolution | |
| wtdbg2 |
Long | Fuzzy Bruijn graph | Fast and memory-efficient; designed for noisy long reads | |
| Shasta |
Long (ONT) | Marker graph | Fast and memory-efficient | |
| Raven |
Long | OLC | Optimized for long uncorrected reads; fast and memory-efficient | |
| Hifiasm |
Long (HiFi) | String graph | High accuracy and repeat resolution | |
| NECAT |
Long (ONT) | OLC | Efficient assembly of noisy long reads | |
| SmartDenovo | Long | OLC | No error correction | |
| NextDenovo |
Long | OLC | Improved accuracy |
| Tool | Category | Input | Core method | Key function | Reference |
|---|---|---|---|---|---|
| Pilon | Short-read polishing | Illumina reads | Read mapping | Error correction of SNPs and small indels | |
| Polypolish | Short-read polishing | Illumina reads | Multi-mapping | Repeat-aware error correction | |
| Pypolca | Short-read polishing | Illumina reads | Read mapping | Error correction with threshold-based variant filtering | |
| Racon | Long-read polishing | Long reads | Read mapping | Consensus-based error correction | |
| NeuralPolish | Long-read polishing | Long reads | Deep learning | Improved base accuracy using neural networks | |
| DeepPolisher | Long-read polishing | Long reads | Deep learning | Deep learning–based error correction | |
| Medaka | Long-read polishing | ONT reads | Deep learning | Signal-aware error correction | |
| NextPolish | Hybrid polishing | Short + long reads | Iterative polishing | Multi-platform error correction |
| Tool | Category | Input | Core method | Key function | Reference |
|---|---|---|---|---|---|
| REAPR | Read-based | Short reads + assembly | Read mapping | Error detection | |
| Inspector | Read-based | Long reads | Read mapping | Structural and local error detection with correction | |
| QUAST | Reference-based | Assembly + reference | Whole-genome alignment | Assembly quality assessment and misassembly detection | |
| Assemblytics | Reference-based | Assembly + reference | Whole-genome alignment | Structural variation detection | |
| CheckM | Completeness | Assembly | Lineage-specific marker genes | Completeness + contamination | |
| BUSCO | Completeness | Assembly | Marker genes (Single-copy orthologs) | Completeness assessment | |
| KAT | k-mer based | Reads + assembly | k-mer comparison | Coverage bias, duplication detection | |
| Merqury | k-mer based | Reads + assembly | k-mer spectrum | Accuracy (QV) + completeness | |
| Bandage | Graph-based | Assembly graph | Visualization | Graph inspection | |
| gfatools | Graph-based | GFA | Graph parsing | Structural analysis | |
| Circlator | Structural | Assembly | Overlap detection | Circularization validation | |
| MOB-suite | Plasmid | Assembly | Database + typing | Plasmid reconstruction |
Tools marked with an asterisk are actively maintained and commonly used in contemporary bacterial genome assembly pipelines.
Table 1.
Table 2.
Table 3.
TOP

