ABSTRACT
- A Gram-stain-negative, aerobic, non-motile, rod-shaped, and orange-pigmented bacterium, designated CJ426T, was isolated from ginseng soil in Anseong, Korea. Strain CJ426T grew optimally on Reasoner’s 2A agar at 30°C and pH 7.0 in the absence of NaCl. Phylogenetic analysis based on the 16S rRNA gene sequence revealed that strain CJ426T belonged to the family Chitinophagaceae and had the highest sequence similarity with Niabella hibiscisoli KACC 18857T (98.7%). The 16S rRNA gene sequence similarities with other members of the genus Niabella ranged from 92.3% to 98.1%. Phylogenomic analyses and overall genomic relatedness indices, including average nucleotide identity, average amino acid identity, and the percentage of conserved proteins values, supported the classification of strain CJ426T as a representative of a novel genus within the family Chitinophagaceae. Furthermore, genome-based analyses suggested that five members of the genus Niabella, including N. aquatica, N. defluvii, N. ginsengisoli, N. hibiscisoli, and, N. yanshanensis, should be separated from other Niabella species and be assigned as a novel genus. The major isoprenoid quinone of strain CJ426T was menaquinone-7 (MK-7). The predominant polar lipids were phosphatidylethanolamine and six unidentified aminolipids. The major fatty acids were iso-C15:0, iso-C15:1 G, and iso-C17:0 3-OH. The genome of strain CJ426T was 6.3 Mbp in size, consisting of three contigs, with a G + C content of 41.9%. Based on a polyphasic taxonomic approach, strain CJ426T represents a novel genus and species within the family Chitinophagaceae, for which the name Paraniabella aurantiaca gen. nov., sp. nov. is proposed. The type strain is CJ426T (= KACC 23908T = JCM 37728T).
-
Keywords: Paraniabella, Paraniabella aurantiaca, genome, reclassification, OGRI
Introduction
The family Chitinophagaceae, belonging to the order Chitinophagales within the phylum Bacteroidota, was initially proposed by Kämpfer et al. (2011) and has been reported in a wide range of habitats, including soil, fresh water, marine, air, plants, humans, and hot springs. The members of the family are characterized as Gram-stain-negative, usually non-motile, rod-shaped, and either aerobic or facultatively anaerobic. At the time of the current writing, the List of Prokaryotic Names with Standing in Nomenclature (LPSN) encompassed 56 validly published names within the family Chitinophagaceae (Parte et al., 2020).
The genus Niabella was first proposed by Kim et al. (2007). Phylogenetically, the genus Niabella belongs to the family Chitinophagaceae in the class Chitinophagia. Currently, LPSN listed 14 validly published names under the genus Niabella (Parte et al., 2020). Members of the genus Niabella have been isolated from diverse sources, including lake water (Siddiqi and Im, 2016), influent water (Min et al., 2024), wetlands (Guo et al., 2022), soil samples (Dahal and Kim, 2016; Dai et al., 2011; Kim et al., 2007; Lin et al., 2022; Ngo et al., 2017; Wang et al., 2009; Weon et al., 2008, 2009), compost (Yi et al., 2015), and medicinal leeches (Hirudo verbena) (Glaeser et al., 2013). The genus is characterized as Gram-stain-negative, short rod-shaped, aerobic, non-flagellated, non-spore-forming, and flexirubin-pigment-producing. The major respiratory quinone is menaquinone-7 (MK-7), and the major fatty acids include iso-C15:0, iso-C15:1 G, and iso-C17:0 3-OH.
In this study, a novel bacterium, designated strain CJ426T, was isolated from ginseng soil and characterized using a polyphasic taxonomic approach in comparison with related type strains. Based on polyphasic taxonomic analyses, strain CJ426T is proposed as a novel species and genus, for which the name Paraniabella aurantiaca gen. nov., sp. nov. is proposed. Furthermore, genome-based phylogenetic analyses suggest that the closely related phylogenetic neighbours N. aquatica, N. defluvii, N. ginsengisoli, N. hibiscisoli, and N. yanshanensis be reclassified into this novel genus.
Materials and Methods
Isolation of bacterial strain and culture conditions
A novel bacterial strain was isolated from ginseng soil in Anseong, Korea (37°02'24.8"N 127°21'58.6"E). The ginseng soil samples (approximately 1–2 g) were suspended in 50 ml of R2A broth (BD) and incubated at 30℃ for 48 h with shaking (200 rpm). The enriched culture was then serially diluted, spread onto R2A agar plates, and incubated at 30℃ for 48 h. Several single colonies were picked and purified by repeated streaking on fresh R2A agar plates. Among them, strain CJ426T was cultivated in R2A broth (BD) at 30℃ for two days and preserved in a 30% (w/v) glycerol suspension at –80℃.
16S rRNA gene-based phylogenetic analysis
The 16S rRNA gene was amplified from a single colony via PCR using AccuPower PCR Premix (Bioneer) and primers pBact27F and pUniv1492R (Frank et al., 2008). Sequencing of the 16S rRNA gene was performed at Biofact (Korea) on an automated DNA analyzer (ABI 3730XL; Applied Biosystems) using primers 27F, 1492R, 518F, and 805R to generate a consensus sequence (Baker et al., 2003). The partial 16S rRNA gene sequence of strain CJ426T was compared with those of related type strains using the EzBioCloud server (Chalita et al., 2024). Multiple sequence alignments were performed using MUSCLE (Edgar, 2004), and phylogenetic trees were constructed using the neighbour-joining (NJ), maximum-likelihood (ML), and maximum-parsimony (MP) methods in MEGA 11 software (Tamura et al., 2021). The NJ phylogeny was constructed using the Jukes–Cantor model (Jukes and Cantor, 1969). For the ML phylogeny, the best substitution model was determined based on the Bayesian Information Criterion using the model test option in MEGA 11, and the Kimura 2-parameter model was applied to construct the ML phylogeny. The MP phylogeny was constructed using the pairwise deletion option. Bootstrap analysis with 1000 replicates was employed to evaluate tree topologies (Felsenstein, 1985).
Genomic DNA extraction, whole genome sequencing, and de novo assembly
Genomic DNA was extracted using the DNeasy Blood & Tissue Kits (QIAGEN, Germany) following the manufacturer’s protocols. The whole genome sequencing of strain CJ426T was conducted at CJ Bioscience, Inc. (Korea) using the PacBio sequencing platform. The genomic DNA library for sequencing was prepared using the SMRTbell® Express Template Preparation Kit (PacBio, Cat No, 100-938-900). Genome assembly was performed with SMRTLink v13.1.0 using the Microbial Assembly protocol (Pacific Biosciences, USA). Contigs were rearranged to start at the dnaA/repA replication origin using Circlator v1.5.5 (Hunt et al., 2015). Reconstruction and typing of bacterial plasmid contigs from draft genome assembly data was used MOB-suite (Robertson and Nash, 2018). The assembled genome was evaluated using BUSCO v5.8.0 with single-copy gene datasets of Bacteroidota lineages v5 (Manni et al., 2021) and CheckM v1.2.3 with the marker lineage “p__Bacteroidetes” following to the default parameters. The assembled genome was visualized using Genovi v0.2.16 (Cumsille et al., 2023).
Genomic and phylogenomic analyses
The genomic analysis adhered to the minimal standards for genome data proposed by Chun et al. (2018). Digital DNA-DNA hybridization (dDDH) values were calculated using the Genome-to-Genome Distance Calculator (GGDC 3.0) (Meier-Kolthoff et al., 2022) with formula 2. Average nucleotide identity (ANI) values were obtained using the OrthoANI pipeline (Lee et al., 2016). In accordance with the updated minimal standards for genome data at the genus level proposed by Riesco and Trujillo (2024), average amino acid identity (AAI) values were determined using the EzAAI pipeline (Kim et al., 2021b), and the percentage of conserved proteins (POCP) for genus-level classification was calculated according to Qin et al. (2014). Reference genome sequences for comparison with strain CJ426T were sourced from the National Center for Biotechnology Information (NCBI) datasets (O'Leary et al., 2024) and the Type Strains Genome database (gcTYPE) (Fan et al., 2024).
Protein-coding genes, rRNA, tRNA, and tmRNA genes were predicted using Prodigal (Hyatt et al., 2010), Infernal (Nawrocki and Eddy, 2013) with bacterial covariance models based on the Pfam database (Kalvari et al., 2021; Schwengers et al., 2021), tRNAscan-SE 2.0 (Chan et al., 2021), and Aragorn (Laslett and Canback, 2004), respectively. Functional annotation was performed using Bakta v1.9.4 (Schwengers et al., 2021), incorporating data from three UniProt Reference Cluster databases (UniProt, 2021) and ten additional databases. The functional metabolic pathways of type strains in the genus Niabella were reconstructed using the KEGG database within METABOLIC v4.0 software (Zhou et al., 2022). Carbohydrate-active enzymes and proteolytic enzymes were annotated with HMMER v3.4 (Finn et al., 2011) using the CAZy database implemented in the dbCAN2 server (Zheng et al., 2023) and the MEROPS database (Rawlings et al., 2017), respectively. Antibiotic resistance genes (ARGs) were identified using the NCBI Antimicrobial Resistance Gene Finder (Feldgarden et al., 2021). Secondary metabolite biosynthetic gene clusters (BGCs) were identified using antiSMASH v7.0 (Blin et al., 2024).
The up-to-date bacterial core-genes 2 (UBCG2) phylogenomic pipeline (Kim et al., 2021a) was used to generate a concatenated alignment of 81 UBCG sequences, and a phylogenomic tree was constructed using IQ-TREE v2.3.5 (Minh et al., 2020) with the transitional (TIM3) substitution model, empirical base frequencies, invariable sites, and four gamma categories. The constructed phylogenomic tree was visualized using MEGA 11 software. The phylogeny produced using UBCGs was compared with trees generated by other methods, including a phylogenomic tree based on 290 concatenated core-gene alignment sequences extracted by Panaroo v1.5.1 (Tonkin-Hill et al., 2020). Alternative trees were also constructed using IQ-TREE v2.3.5 with the general time reversible substitution model, empirical base frequencies, invariable sites, and four gamma categories.
The pan-genomes of type strains in the genus Niabella were calculated using Panaroo v1.5.1, with Bakta-annotated genomes as input. A protein sequence identity threshold of 90% (--core_threshold 0.90) and a length difference cutoff of 98% (--len_dif_percent 0.98) were applied in strict mode (--clean-mode strict). The protein family sequence identity threshold was set to 50% (-f 0.5). The assembled genome was compared with the reference genomes of related type strains through whole-genome alignment using NUCmer v3.23 (Kurtz et al., 2004) and visualized with Circos v0.69.8 (Krzywinski et al., 2009) implemented via Mummer2Circos (https://github.com/metagenlab/mummer2circos).
Morphological and physiological characterization
The cellular morphology of strain CJ426T was observed using transmission electron microscopy. Cells were grown for two days at 30℃ on R2A agar. Growth was assessed on various agar media, including R2A agar, tryptic soy agar (TSA), Luria-Bertani (LB) agar, nutrient agar (NA), and marine agar (MA). To evaluate growth temperatures, cells were cultured on R2A agar at 4, 10, 15, 20, 25, 30, and 37℃. The pH growth range was determined using 0.1 M citrate buffer (pH 4.0–5.0), 0.1 M phosphate buffer (pH 6.0–8.0), and 0.1 M bicarbonate–carbonate buffer (pH 9.0–10.0), 0.05 M disodium hydrogen phosphate-sodium hydroxide buffer (pH 11.0–12.0), and 0.2 M potassium phosphate-sodium hydroxide buffer (pH 13.0) in R2A broth (Bates and Bower, 1956). Salt tolerance was tested by supplementing R2A broth with 0–3% (w/v) NaCl (at 1% intervals). Both the NaCl tolerance and pH range experiments were conducted at 30℃ for up to 3 days. Gram-staining was performed using a Gram-staining kit according to the manufacturer’s protocols (Sigma-Aldrich). Gliding motility was examined on semisolid R2A media containing 0.4% agar. Anaerobic growth was determined after two weeks of incubation at 30℃ on R2A agar using the GasPak Anaerobe Pouch System (BD). The production of flexirubin-type pigments was tested using a 20% KOH test and interpreted by changes in colony color. Oxidase and catalase activities were determined using oxidase reagent (bioMerieux) and a 3% (v/v) aqueous H2O2 solution, respectively. Hydrolysis of starch, cellulose, casein, and DNA was tested using media containing 1.0% (w/v) soluble starch, 0.2% (w/v) carboxymethyl (CM) cellulose, 3% (w/v) skimmed milk, and DNase test agar (BD), respectively. Results of the hydrolysis tests were evaluated after 2 days of incubation. Carbon source utilization and enzymatic activities of strain CJ426T were determined using the API 20NE according to the manufacturer’s instructions.
Chemotaxonomic analyses
The polar lipids of strain CJ426T were extracted from 100 mg of freeze-dried cells cultured in R2A agar for two days at 30℃ and analyzed by two-dimensional silica gel thin-layer chromatography (TLC) (Tindall, 1990a, 1990b). Total lipids and specific functional group lipids were detected using molybdophosphoric acid, molybdenum blue spray, ninhydrin reagent and α-naphthol. The respiratory quinone was extracted as described by Minnikin et al. (1984) and determined by high performance liquid chromatography (HPLC) analysis using a reverse phase column (Apollo C18, 250 × 4.6 mm, 5 μm) (Collins, 1985). The quinones were eluted by a mixture of methanol and 2-propanol (4:1, v/v) with a flow rate of 1 ml/min and detected by UV absorbance at 270 nm. For fatty acid analysis, strain CJ426T and related type strains were cultivated in R2A broth at 30℃ and harvested at the exponential growth phase. Fatty acids were saponified, methylated, and extracted following the standard protocol of the Sherlock Microbial Identification System (MIDI) version 6.2 and analyzed by the standard protocol of gas chromatography (Agilent Technologies). The cellular fatty acids were identified using the RTSBA6 database of the Microbial Identification System (Sasser, 1990).
Antimicrobial susceptibility test
The antibiotic susceptibility of strain CJ426T was evaluated using the disk diffusion assay with antibiotic impregnated disks (Lioflchem, Italy). The following antibiotic were used: amoxicillin (10 μg), cephalexin (30 μg), chloramphenicol (10 μg), ciprofloxacin (5 μg), clindamycin (2 μg), colistin (10 μg), erythromycin (10 μg), fosfomycin (200 μg), gentamicin (10 μg), linezolid (10 μg), meropenem (10 μg), rifampicin (5 μg), streptomycin (10 μg), sulfamethoxazole (50 μg), tetracycline (30 μg), trimethoprim (5 μg), tylosin (30 μg), vancomycin (5 μg). The susceptibility results were recorded as susceptible (S), intermediate (I), or resistant (R) based on the breakpoints for Enterobacterales and Staphylococcus spp. according to the CLSI supplement M100 (CLSI, 2025).
Statistical analyses and visualization
Differences in genome size and G + C contents between the genera Niabella and Paraniabella were assessed using the t-test in R. A flower plot was visualized in R using the ‘plotrix’ packages. Scatter plot and box plots were generated in R using the ‘ggplot2’ package. The overall genomic relatedness indices (OGRI) heatmaps were visualized in R using the ‘pheatmap’ package. Pairwise correlations between ANI, AAI, and POCP values were calculated and visualized in R with ‘psych’ package. Functional annotation results on phylogenetic trees were visualized in R using the ‘ggplot2’, ‘ggtree’, ‘aplot’, ‘ggstance’, ‘patchwork’, and ‘ggstar’ packages.
Nucleotide sequence accession numbers
The EMBL/DDBJ/GenBank accession numbers for the 16S rRNA gene sequence and genome sequence of strain CJ426T are KF740309 and JBLOSY000000000, respectively.
Results and Discussion
Phylogeny based on 16S rRNA gene and whole genome sequences
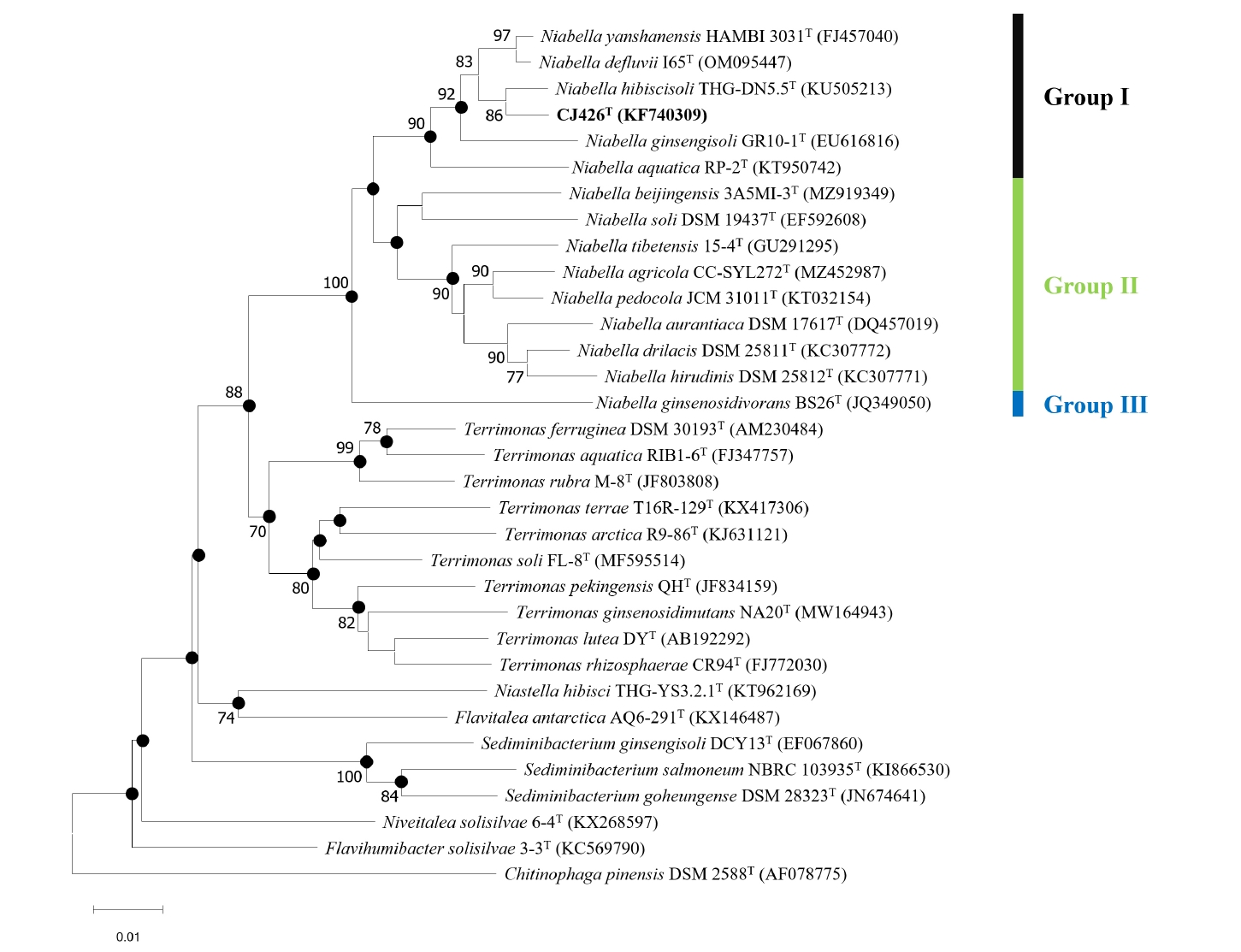
The 16S rRNA gene sequence of strain CJ426T showed the highest similarity (98.7%) to N. hibiscisoli THG-DN5.5T, followed by N. yanshanensis KACC 14980T (98.1%), and N. defluvii I65T (97.8%). In contrast, strain CJ426T showed the lowest similarity (92.3%) to N. tibetensis 15-4T, followed by N. beijingensis 3A5MI-3T (93.2%), and N. agricola CC-SYL272T (93.2%). The pairwise 16S rRNA gene sequence identities, which are below the established thresholds for species (≤ 98.7%) (Kim et al., 2014) and genus delineation (≤ 94.5%; 95% confidence interval [CI]: 94.55–95.05%) (Yarza et al., 2014), suggest that strain CJ426T can be proposed as a novel species and genus within the family Chitinophagaceae (Tables S1 and S2). Furthermore, based on the 16S rRNA gene similarity and the phylogenetic trees, the genus Niabella was divided into three preliminary groups (Fig. 1). According to the phylogenetic trees (Figs. 1, S1, and S2), strain CJ426T formed a robust cluster with N. hibiscisoli THG-DN5.5T and constructed a large clade with N. aquatica RP-2T, N. ginsengisoli GR10-1T, N. defluvii I65T, and N. yanshanensis HAMBI 3031T (named Group Ⅰ). The remaining Niabella species formed another distinct clade (named Group Ⅱ) separated from the above species (Group Ⅰ), while N. ginsenosidivorans BS26T formed a single distinct lineage (named Group Ⅲ). To elucidate more accurate taxonomic relationships among these groups, comprehensive analyses were performed using whole genome sequence data.
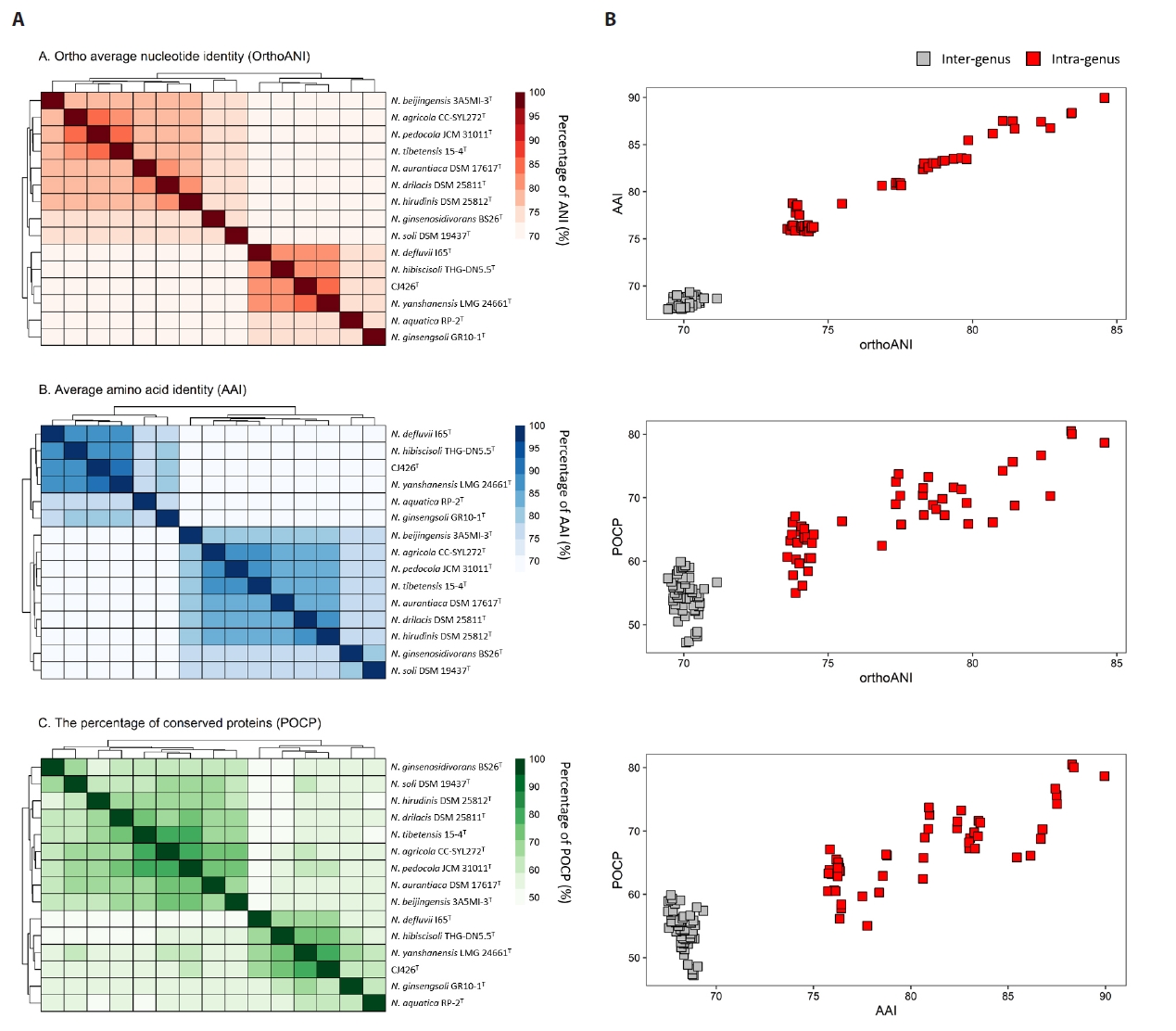
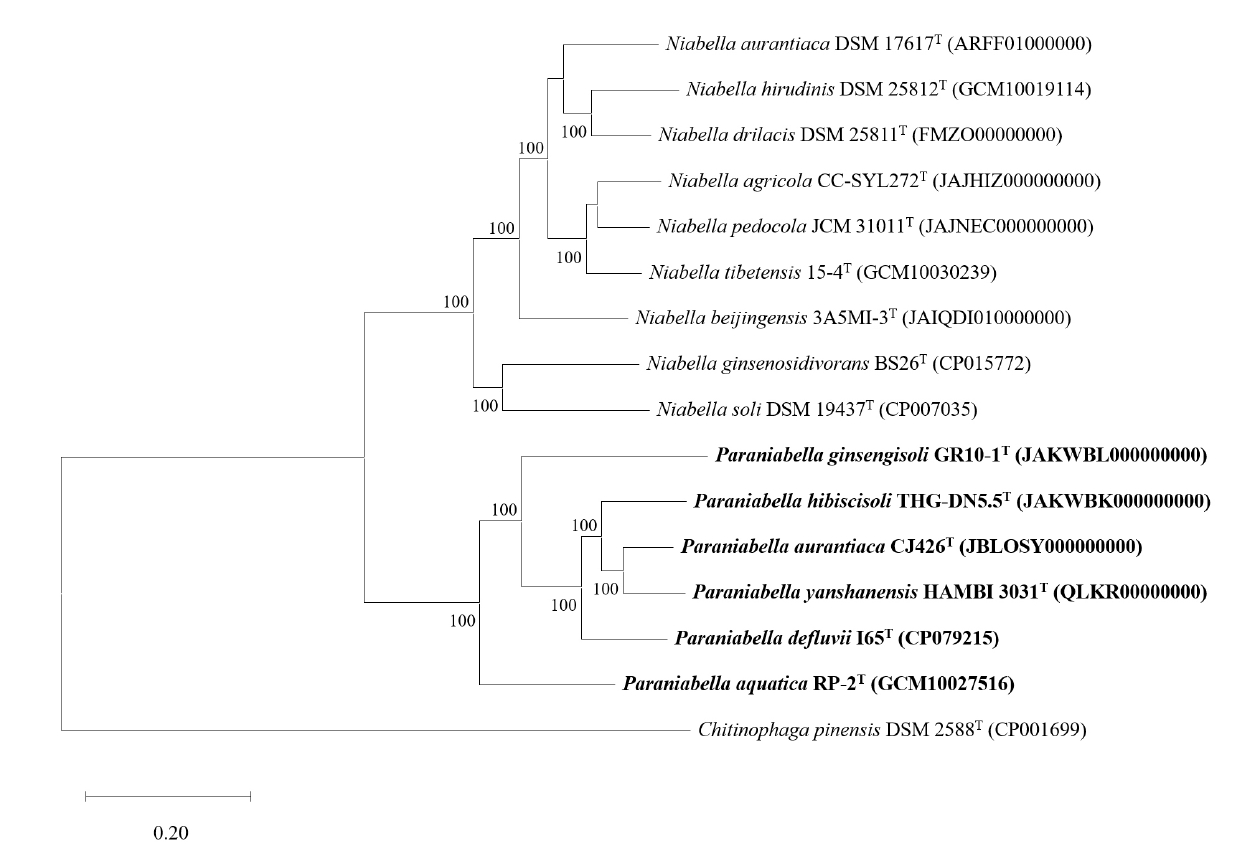
To analyze the overall genomic similarity between strain CJ426T and its related type strains, ANI, AAI, dDDH, and POCP indices were calculated (Fig. 2A, Table S3). The ANI values between strain CJ426T and all related type strains in the genus Niabella were 69.9% to 84.6% (Table S3). The dDDH values between strain CJ426T and the related type strains were all below 30%. According to established thresholds, the ANI values below 95–96% and the dDDH values below 70% clearly suggest that strain CJ426ᵀ represents a novel species within the family Chitinophagaceae at the genomic level (Chun et al., 2018; Meier-Kolthoff et al., 2013; Richter and Rosselló-Móra, 2009). For the genus delineation, the intra-group and inter-group ANI values were evaluated (Table 1). The intra-group ANI values within Group Ⅰ and Group Ⅱ were 73.6–84.6% and 73.7% to 83.4%, respectively. In contrast, the inter-group ANI values were 69.5–70.7% between Group Ⅰ and Group Ⅱ, 70.0–71.2% between Group Ⅰ and Group Ⅲ, and 74.2–75.5% between Group Ⅱ and Group Ⅲ. The ANI value range between Group Ⅰ and the other groups (Group Ⅱ and Group Ⅲ) was 69.5–71.2%, which was not only lower than the intra-group ANI values but also below the recently proposed genus boundary (an average of 73.10%, 95% CI: 72.50% to 73.70%) (Barco et al., 2020), suggesting that Group Ⅰ can be separated from the other groups at the genomic level. Additionally, AAI and POCP values were also used for genus delineation (Tables S3 and S4). The intra-group AAI values within Group Ⅰ and Group Ⅱ were 76.1–90.0% and 75.8–88.4%, respectively. In contrast, the inter-group AAI values were 67.5–69.3% between Group Ⅰ and Group Ⅱ, 67.9–68.9% between Group Ⅰ and Group Ⅲ, and 75.7–78.7% between Group Ⅱ and Group Ⅲ. These ranges were not only lower than those of the intra-group AAI values but also falls within the proposed threshold for genus boundary (an average of 70%) (Luo et al., 2014). The intra-group POCP values within Group Ⅰ and Group Ⅱ were 55.0–78.7% and 62.4–80.5%, respectively. In contrast, the inter-group AAI values were 47.2–59.9% between Group Ⅰ and Group Ⅱ, 48.2–56.7% between Group Ⅰ and Group Ⅲ, and 58.4–66.3% between Group Ⅱ and Group Ⅲ. Consequently, the POCP values between the members of Group Ⅰ and those of the other groups (Group Ⅱ and Group Ⅲ) had a lower range (47.2–59.9%) than the intra-group POCP values. Recent studies reported that the genera Cnuella and Paracnuella within the family Chitinophagaceae were separated with an inter-genus POCP value of 69.0% (Yuan et al., 2023). Meanwhile, pairwise comparisons of ANI, AAI, and POCP values indicated that the pair of ANI and AAI had a higher correlation coefficient (Spearman’s ρ = 0.88, p < 0.001) than other pairs (Fig. S3). Consequently, the results from ANI, AAI, and POCP analyses support that the members of Group Ⅰ are separable from the other groups (Fig. 2B). The relatively higher inter-group ANI, AAI, and POCP values between Group Ⅱ and Group Ⅲ, compared to those between Group I and the other groups, also suggest that Group Ⅱ and Group Ⅲ should be considered as a single group representing the genus Niabella. Based on genome-based analyses, the genus Niabella is highly divergent, highlighting the need for reclassification of the genus Niabella. Furthermore, the two phylogenomic trees showed that members of Group Ⅰ formed a distinct phylogenetic lineage separated from the other members of the genus Niabella (Figs. 3 and S4). Therefore, we propose that strain CJ426T and closely related type strains within Group Ⅰ should be classified as the novel genus Paraniabella.
Genomic features
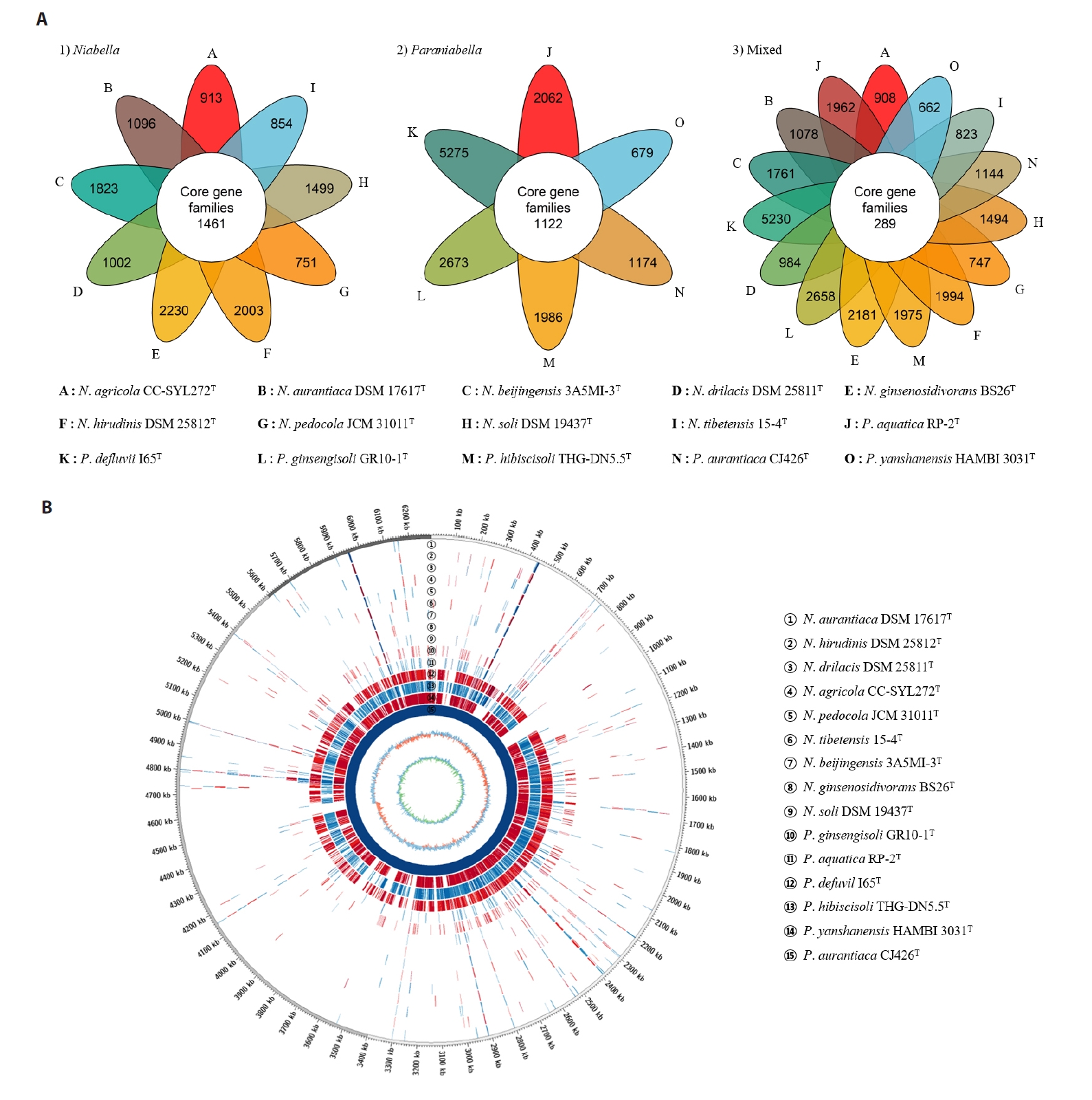
The draft genome of strain CJ426ᵀ comprised three contigs with no plasmids detected and had a total size of 6.3 Mbp (Fig. S5). The completeness of the genome assembly was evaluated to be 97.5% using BUSCO and 99.5% with a contamination level of 0.5% using CheckM. The genomic DNA G + C content of strain CJ426T was calculated to be 41.9% based on genome sequence. The genome sequence of strain CJ426T contained 5,310 CDSs, 45 tRNA genes, and 6 rRNA genes, including two identical copies of the 16S rRNA gene. A summary of the general genomic features of strain CJ426ᵀ and related type strains is provided in Table 2. Comparison of general genomic features between the two genera revealed a significant difference in G + C contents (p < 0.001) but no significant difference in genome size (Fig. S6). The differences in G + C content values between species of the two genera Niabella (45.2–48.6%, an average of 46.8%) and Paraniabella (38.5–43.0%, an average of 41.7%) exceeded the cut-off of ≥ 1% within genus, also suggesting that strain CJ426T and its related type strains should be classified as a novel genus, distinct from other Niabella type strains (Meier-Kolthoff et al., 2014). Pangenome analysis revealed that the genus Niabella and the proposed genus Paraniabella possessed 1,461 and 1,122 core orthologous genes, respectively (Fig. 4A). However, when all members of both genera were analyzed together, the number of core orthologous genes sharply decreased to 289 (Fig. 4A), further justifying the separation of these two genera. The whole genome sequence alignment analysis showed that strain CJ426T and its related type strains within the genus Paraniabella exhibited higher similarities compared to other type strains within the genus Niabella (Fig. 4B).
According to functional annotation based on KEGG pathway, strain CJ426T possessed 64 complete metabolic pathway modules (Fig. S7A), including well-conserved central carbohydrate metabolism pathways such as glycolysis, the Entner-Doudoroff pathway, pyruvate oxidation, gluconeogenesis, the citrate cycle, phosphoribosyl pyrophosphate biosynthesis and the pentose phosphate pathway (Table S4). Moreover, biosynthetic module for tryptophan was identified and consistent with phenotype production of indole. A single aminoglycoside O-nucleotidyltransferase (aadS;ant(6)), rifampin ADP-ribosyltransferase (arr), and two β-lactamase genes (subclass A2 and B3), which confer resistance to aminoglycosides, rifampin, and certain β-lactam antibiotics, were predicted in the genome sequence of strain CJ426T (Fig. S7B). Strain CJ426ᵀ encoded the highest number of antibiotic resistance genes compared to other related type strains: subclass A2 β-lactamase gene is exclusively present in its genome, which is known to hydrolyze penicillins and older cephalosporins (CfxA, CIA-1, CME-1, PER-1, and VEB-1) (Philippon et al., 2016). These results were consistent with phenotypic resistance to amoxicillin, meropenem, rifampicin and streptomycin (Table S5). Annotation of carbohydrate-active and proteolytic enzymes revealed that the genome of strain CJ426ᵀ contained 239 carbohydrate-active enzymes, which were classified into two distinct families of CAZymes: 220 glycoside hydrolases and 19 polysaccharide lyases (Fig. S7B and Table S6). Additionally, the genome of strain CJ426T encoded 62 proteolytic enzymes, including one aspartic peptidase, seven cysteine peptidases, one glutamic peptidase, 23 metallo peptidases, 29 serine peptidases, and one threonine peptidase (Fig. S7B and Table S7). Furthermore, the genome of strain CJ426T contained several biosynthetic gene clusters (BGCs), including one lasso peptide BGC (8% similarity to known gene clusters), two copies of terpene BGCs (one showing 28% similarity to a known carotenoid BGC), two copies of flexirubin BGC (44% and 28% similarity to known gene clusters, respectively), one non-ribosomal peptide synthase (NRPS) BGC, and one type Ⅲ polyketide synthase (PKS) BGC. A summary of the BGCs of strain CJ426ᵀ is provided in Table S8.
Morphological, physiological, and biochemical characteristics
Cellular morphology was shown to be rod-shaped and non-flagellated (Fig. S7). Optimal growth media was R2A medium, but growth also occurred on TSA, NA, LB, and MA medium. Growth of strain CJ426T occurred at 4–37℃ (optimum 30℃), at pH 5.0–12.0 (optimum pH 7.0), and at 0–2% concentration of NaCl (optimum 0%). Strain CJ426T produced flexirubin-like pigments. The differential biochemical and physiological characteristics of strain CJ426T and the type strains of related genera within the family Chitinophagaceae are summarized in Table 3. Additionally, differential characteristics among the members of the genera Niabella and Paraniabella are provided in Table S9. The genera Niabella and Paraniabella exhibit highly similar morphological, physiological and biochemical characteristics. Although phenotypic traits were not clearly distinguished between the two genera, all members of the proposed genus Paraniabella shared a consistent set of characteristics that differ from those of Niabella. For instance, all members of Paraniabella are positive for catalase activity and can assimilate D-glucose, L-arabinose, D-mannose, and D-maltose, but are unable to assimilate D-mannitol. In contrast, these traits are variable within the genus Niabella.
Chemotaxonomic characteristics
The polar lipids of strain CJ426T were identified as phosphatidylethanolamine, six unidentified aminolipids, and two unidentified lipids (Fig. S9). The respiratory quinone of strain CJ426T was identified exclusively as menaquinone 7 (MK-7). The major fatty acids of strain CJ426T included iso-C15:0 (38.2%), iso-C15:0 G (20.2%), and iso-C17:0 3-OH (14.0%). As shown in Table 4, the fatty acid profile of strain CJ426T was similar to those of closely related type strains within the family Chitinophagaceae, sharing major fatty acid components. These chemotaxonomic characteristics are consistent with those of other recognized species in the family Chitinophagaceae. The fatty acid profiles of strain CJ426T and closely related type strains of the genera Niabella and Paraniabella are summarized in Table S10. Furthermore, a comparison of chemotaxonomic characteristics revealed that the genera Niabella and Paraniabella have similar fatty acid profiles, with the main difference being that the percentage of summed feature 3 tends to be higher in members of Niabella than in those of Paraniabella.
Taxonomic conclusion
Phylogenetic analyses indicated that strain CJ426T and its closely related type strains, N. aquatica, N. defluvii, N. ginsengisoli, N. hibiscisoli, and N. yanshanensis, formed distinct clades within the genus Niabella in all reconstructed trees, including NJ, ML, and MP phylogenetic trees based on the 16S rRNA gene sequences, as well as phylogenomic trees based on UBCGs and 290 SCGs concatenated alignment sequences, repectively. Moreover, the low OGRI values (ANI, AAI, POCP, and dDDH) bewteen the proposed Paraniabella group (including strain CJ426T) and the remaining members of the genus Niabella strongly support the reclassification of this group as a representative of a novel genus. Significant differences in G + C contents and core orthologous genes between these two groups also support the genus delineation. Based on polyphsic taxonomy study, strain CJ426T is distinguished from closely related type strains on the basis of genome, chemical, molecular, and physiological characteristics, including metabolic pathways, antibiotic resistance genes, carbon source utilization and growth ranges. Futhermore, chemotaxonomic and phenotypic characteristics, such as respiratory quinone, polar lipid, fatty acid, and carbon source utilization, were concordant with the traits listed in the family Chitinophagaceae description.
Therefore, strain CJ426T represents a novel species of a novel genus within the family Chitinophagaceae, for which the name Paraniabella aurantiaca gen. nov., sp. nov. is proposed. In addition, we propose that its phylogenetic neighbours N. aquatica, N. defluvii, N. ginsengisoli, N. hibiscisoli, and N. yanshanensis be reclassified into this novel genus as Paraniabella aquatica comb. nov., Paraniabella defluvii comb. nov., Paraniabella ginsengisoli comb. nov., Paraniabella hibiscisoli comb. nov., and Paraniabella yanshanensis comb. nov., respectively. On the basis of the genomic data obtained in this study, the description of the genus Niabella is emended.
Description of Paraniabella gen. nov.
Paraniabella (Gr. prep. para, beside, near, like; N.L. fem. n. Niabella, a bacterial genus; N.L. fem. n. Paraniabella, a genus like Niabella).
Cells are Gram-negative, aerobic, non-motile, rod-shaped. Catalase activity is positive. D-Glucose, L-arabinose, D-mannose, and D-maltose assimilated, but D-mannitol is not. The major fatty acid (> 10%) are iso-C15:0, iso-C15:1 G, iso-C17:0 3-OH, and the isoprenoid quinone is MK-7. A common major polar lipid is phosphatidylethanolamine. The genomic DNA G + C contents are 38.5–43.0%. The type species is Paraniabella ginsengsoli. The genus belongs to the family Chitinophagaceae.
Description of Paraniabella aurantiaca sp. nov.
Paraniabella aurantiaca (au.ran.ti'a.ca. N.L. fem. adj. aurantiaca, orange-coloured).
Cells are Gram-negative, aerobic, non-motile, rod-shaped (1.5 µm long and 0.6 µm wide. Colonies on R2A agar are circular, opaque, orange-coloured. Growth occurs at 4–37°C (optimum 30°C), at pH 5.0–12.0 (optimum pH 7.0) and with 0–2% NaCl (optimum 0%). Flexirubin-type pigments are produced. Did not show any motility on 0.4% agar. Casein, cellulose, gelatin, esculin ferric citrate and starch are hydrolyzed, but DNA is not. Positive for catalase but negative for oxidase. Nitrate and nitrite are not reduced. Indole production is detected. D-Glucose is not fermented. Enzyme activities for arginine dihydrolase and β-galactosidase are positive, but urease is not. According to the API 20NE test, D-glucose, L-arabinose, D-mannose, N-acetyl-glucosamine, and D-maltose are assimilated, but D-mannitol, potassium gluconate, capric acid, adipic acid, malic acid, trisodium citrate and phenylacetic acid are not. The predominant polar lipid is phosphatidylethanolamine. The major fatty acid is iso-C15:0, iso-C15:1 G, and iso-C17:0 3-OH. The isoprenoid quinone is MK-7. The type strain is CJ426T (= KACC 23908T = JCM 37728T), isolated from ginseng soil in Korea. The genomic DNA G + C content of the type strain is 41.9%. The EMBL/DDBJ/GenBank accession numbers for the 16S rRNA gene sequence and the whole genome sequence of strain CJ426T are KF740309 and JBLOSY000000000, respectively.
Description of Paraniabella aquatica comb. nov.
Paraniabella aquatica (a.qua′ti.ca. L. fem. adj. aquatica aquatic, from water).
Basonym: Niabella aquatica Siddiqi and Im. 2016
The description is identical to that given for N. aquatica (Siddiqi and Im, 2016). Phylogenomic analysis and overall genome relatedness indices strongly support the placement of this species within the novel genus Paraniabella. The type strain is RP-2T (= KACC 18623T = JCM 30952T).
Description of Paraniabella defluvii comb. nov.
Paraniabella defluvii (de. flu′vi.i. L. gen. n. defluvii, of sewage).
Basonym: Niabella defluvii Min et al. 2024
The description is identical to that given for N. defluvii (Min et al., 2024). Phylogenomic analysis and overall genome relatedness indices strongly support the placement of this species within the novel genus Paraniabella. The type strain is I65T (= KACC 22647T = JCM 35315T).
Description of Paraniabella ginsengisoli comb. nov.
Paraniabella ginsengisoli (gin.seng.i.so′li. N.L. n. ginsengum ginseng; L. n. solum soil; N.L. gen. n. ginsengisoli of soil from a ginseng field, the source of the type strain).
Basonym: Niabella ginsengisoli Weon et al. 2009
The description is identical to that given for N. ginsengisoli (Weon et al., 2009). Phylogenomic analysis and overall genome relatedness indices strongly support the placement of this species within the novel genus Paraniabella. The type strain is GR10-1T (= KACC 13021T = JCM 15444T).
Description of Paraniabella hibiscisoli comb. nov.
Paraniabella hibiscicoli (hi.bis.ci.so′li. N.L. n. hibiscus Rose of Sharon/ Hibiscus syriacus; L. n. solum soil; N.L. gen. n. hibis cisoli of soil of a Rose of Sharon garden, the source of the type strain).
Basonym: Niabella hibiscisoli Ngo et al. 2017
The description is identical to that given for N. hibiscisoli (Ngo et al., 2017). Phylogenomic analysis and overall genome relatedness indices strongly support the placement of this species within the novel genus Paraniabella. The type strain is THG-DN5.5T (= KACC 18857T = CCTCC AB 2016086T).
Description of Paraniabella yanshanensis comb. nov.
Paraniabella yanshanensis (yan.sha.nen′sis. N.L. fem. adj. yanshanensis of Yanshan, an emblem for Hebei Province, where the type strain was isolated).
Basonym: Niabella yanshanensis Wang et al. 2009
The description is identical to that given for N. yanshanensis (Wang et al., 2009). Phylogenomic analysis and overall genome relatedness indices strongly support the placement of this species within the novel genus Paraniabella. The type strain is CCBAU 05354T (= LMG 24661T = HAMBI 3031T).
Emended description of the genus NiabellaDai et al. 2011
The description of the genus is as given by Dai et al. (2011), with the following modifications. Based on the genome sequences, the DNA G + C content of the members of the genus Niabella ranges from 45.2 to 48.6%.
Acknowledgments
We thank Prof. Aharon Oren for his help with the nomenclature. We thank J. Kim for technical help at the BT research facility center, Chung-Ang University. This work was supported by the National Institute of Biological Resources funded by the Ministry of Environment (No. NIBR202502203) and the National Research Foun dation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2023R1A2C1003654).
Author Contributions
Chang-Jun Cha and Yong-Seok Kim designed the study. Material preparation, data collection and analysis were performed by Yong-Seok Kim, Yerang Yang, Miryung Kim, and Do-Hoon Lee. The first draft of the manuscript was written by Yong-Seok Kim, and all authors revised the manuscript. All authors read and approved of the final manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Ethical Statements
Not applicable.
Supplementary Information
The online version contains supplementary material available at https://doi.org/10.71150/jm.2505005.
Table S7.
The number of predicted family domains of proteolytic enzymes based on the MEROPS database for CJ426T and related type strains. -, not detected
jm-2505005-Supplementary-Table-S7.pdf
Table S9.
Characteristics of strain CJ426T and the closely related type strains within the genera Niabella and Paraniabella. Strains: 1, CJ426T; 2, P. hibiscisoli THG-DN5.5T; 3, P. yanshanensis HAMBI 3031T; 4, P. defluvii I65T; 5, P. ginsengisoli GR10-1T; 6, P. aquatica RP-2T; 7, N. beijingensis 3A5MI-3T; 8, N. soli DSM 19437T; 9, N. tibetensis 15-4T; 10, N. agricola CC-SYL272T; 11, N. pedocola JCM 31011T; 12, N. aurantiaca DSM 17617T; 13, N. drilacis DSM 25811T; 14, N. hirudinis DSM 25812T; 15, N. ginsenosidivorans BS26T. The data for strain CJ426 was obtained from this study, while the data for other strains were taken from published literature. +, Positive; w, weakly positive; -, negative; ND, data not available
jm-2505005-Supplementary-Table-S9.pdf
Table S10.
Fatty acid composition of strain CJ426T and related type strains. Strains: 1, CJ426T; 2, P. hibiscisoli THG-DN5.5T; 3, P. yanshanensis HAMBI 3031T; 4, P. defluvii I65T; 5, P. ginsengisoli GR10-1T; 6, P. aquatica RP-2T; 7, N. beijingensis 3A5MI-3T; 8, N. soli DSM 19437T; 9, N. tibetensis 15-4T; 10, N. agricola CC-SYL272T; 11, N. pedocola JCM 31011T; 12, N. aurantiaca DSM 17617T; 13, N. drilacis DSM 25811T; 14, N. hirudinis DSM 25812T; 15, N. ginsenosidivorans BS26T. -, Fatty acid amounts < 1.0% or not detected. The data for strain CJ426 was obtained from this study, while the data for other strains were taken from published literature.
jm-2505005-Supplementary-Table-S10.pdf
Fig. S1.
Maximum-likelihood phylogenetic tree of strain CJ426T and related type strains based on 16S rRNA gene sequences. Bootstrap values greater than 70% are shown at branch points based on maximum-likelihood analysis of 1,000 replicated datasets. Chitinophaga pinensis DSM 2588T was used as an outgroup. Bars, 0.050 substitutions per nucleotide position.
jm-2505005-Supplementary-Fig-S1.pdf
Fig. S2.
Maximum-parsimony (MP) tree based on 16S rRNA gene sequences showing the phylogenetic relationships between strain CJ426T and related type strains of the Genera. Bootstrap values greater than 70% are shown at branch points based on 1,000 replicated datasets. Chitinophaga pinensis DSM 2588T was used as an outgroup.
jm-2505005-Supplementary-Fig-S2.pdf
Fig. S3.
Pairwise correlations of OrthoANI, AAI, and POCP calculated from the genomes of Niabella and Paraniabella type strains. The pairwise correlation calculated using Spearman’s Rank Correlation Coefficient. ***p < 0.001.
jm-2505005-Supplementary-Fig-S3.pdf
Fig. S4.
Phylogenomic tree of strain CJ426T and related type strains based on the concatenated sequences of 290 core genes. Bootstrap values are shown at branch points based on analyses of 1,000 replicated datasets. Chitinophaga pinensis DSM 2588T was used as an outgroup.
jm-2505005-Supplementary-Fig-S4.pdf
Fig. S5.
The draft genome map of Paraniabella aurantiaca CJ426T. From inner to outer tracks: The first track shows the GC skew values with dark green indicating lower than zero and green indicating higher than zero. The second track represents the GC con tent. The third, fourth and fifth tracks display the locations of COG, CDS, tRNA and rRNA on reverse strand of the genome, respectively. The sixth, seventh, and eighth tracks show the locations of tRNA, rRNA, CDS, and COG on forward strand of the genome, respectively.
jm-2505005-Supplementary-Fig-S5.pdf
Fig. S6.
Scatter and box plots for comparison of genome size (Mbp) and G + C content (%) between the genera Paraniabella and Niabella. Statistical significance was assessed using a t-test (NS, not significant; ***p < 0.001).
jm-2505005-Supplementary-Fig-S6.pdf
Fig. S7.
Functional genomic analysis of strain CJ426ᵀ and related type strains. (A) Heatmap showed the presence or absence of metabolic pathway modules based on KEGG annotations. (B) Comparative analysis of functional genes among the type strains based on various databases. The heatmap showed the presence or absence of antibiotic resistance genes. The Bar plot represents the distribution of proteolytic enzymes and CAZYmes. The dendrogram was a phylogenomic tree based on 81 UBCG.
jm-2505005-Supplementary-Fig-S7.pdf
Fig. S9.
Two-dimensional thin-layer chromatography of the polar lipids of strain CJ426T. First dimension, chloroform/methanol/water (65:25:4); second dimension, chloroform/methanol/acetic acid/water (80:12:15:4). Abbreviations: PE, phosphatidylethanolamine; AL, amino lipid; L, unidentified lipid.
jm-2505005-Supplementary-Fig-S9.pdf
Fig. 1.Neighbor-joining tree of strain CJ426T and related type strains based on 16S rRNA gene sequences. Closed circles indicate nodes recovered in the three trees generated by neighbor-joining, maximum-likelihood and maximum-parsimony methods. Bootstrap values greater than 70% are shown at branch points based on 1,000 replicated datasets. Chitinophaga pinensis DSM 2588T was used as an outgroup. Bar, 0.01 substitutions per nucleotide position.
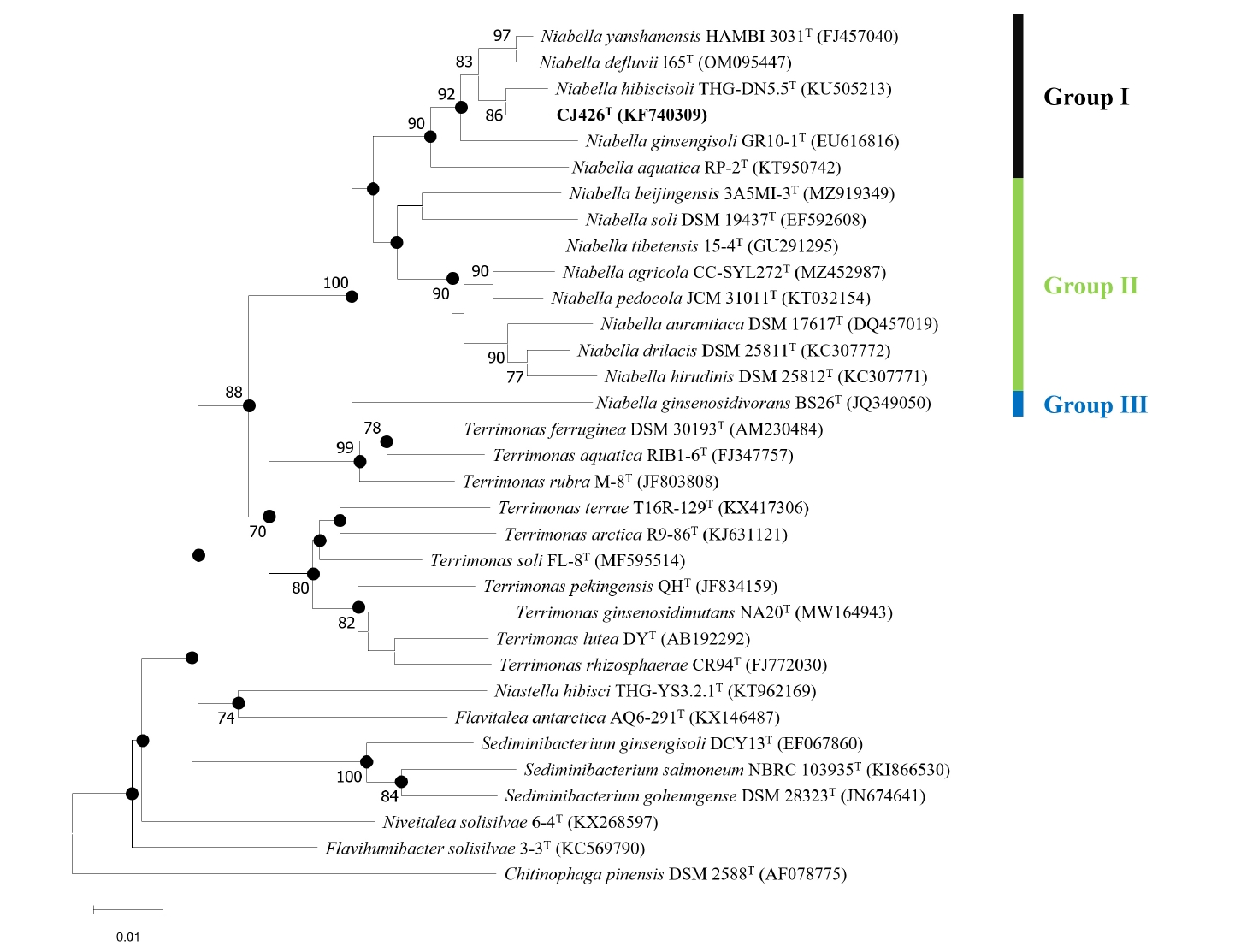
Fig. 2.Comparative genomic features and overall genomic relatedness indices (OGRIs) of strain CJ426T and related type strains. (A) The heatmaps displayed OrthoANI (red), AAI (blue), and POCP (green) values among genomes of the genera Paraniabella and Niabella. Dendrograms indicated clustering based on similarity metrics. Color gradients represented the percentage identity, with darker shades indicating higher similarity. (B) Pairwise correlation plots of OGRI values with proposed taxonomic thresholds for genus delineation.
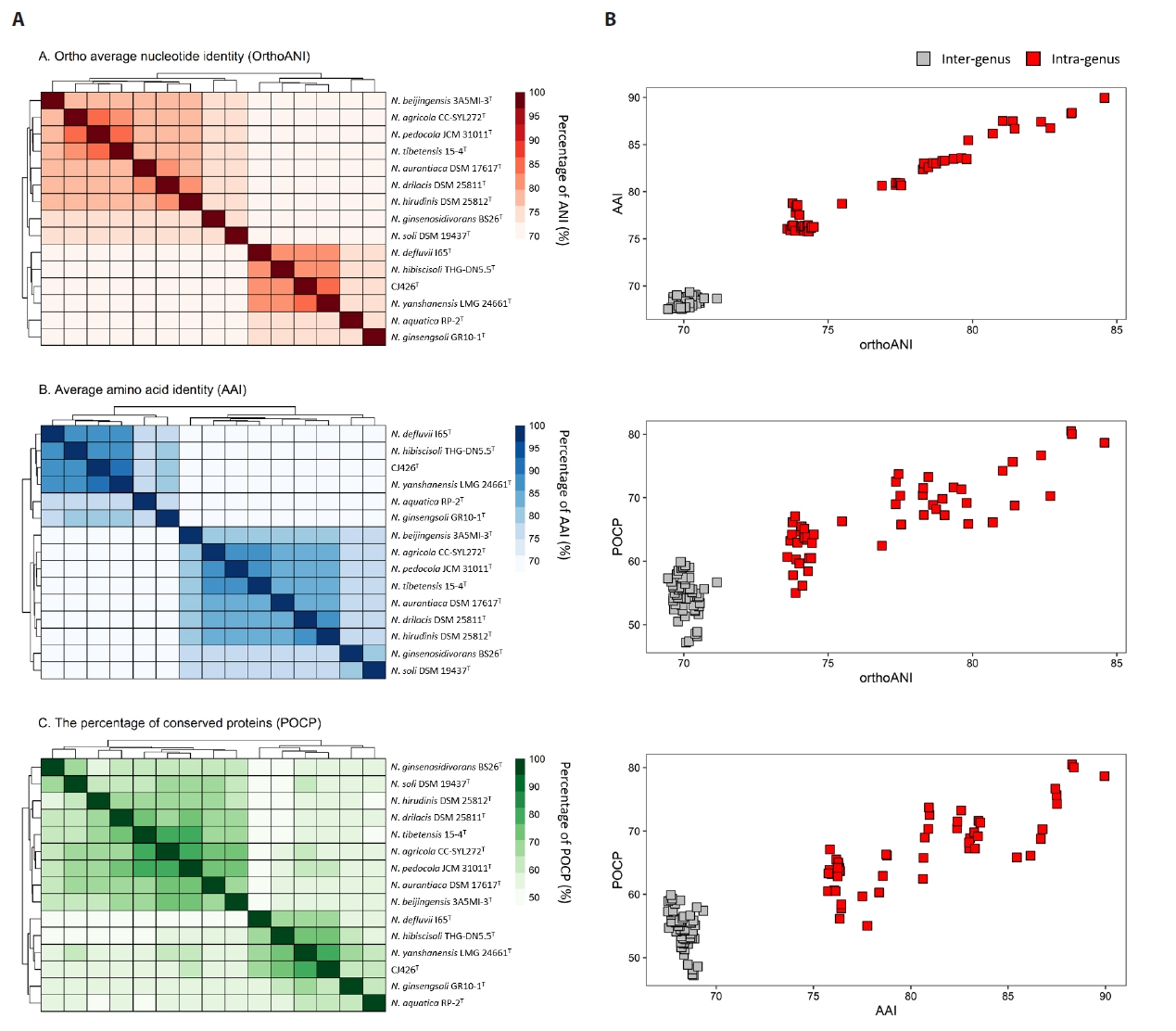
Fig. 3.Phylogenomic tree of strain CJ426T and related type strains based on 81 UBCGs. Bootstrap value greater than 70% are shown at branch points based on maximum-likelihood analysis of 1,000 replicated datasets. Chitinophaga pinensis DSM 2588T was used as an outgroup. Bar, 0.01 substitutions per nucleotide position.
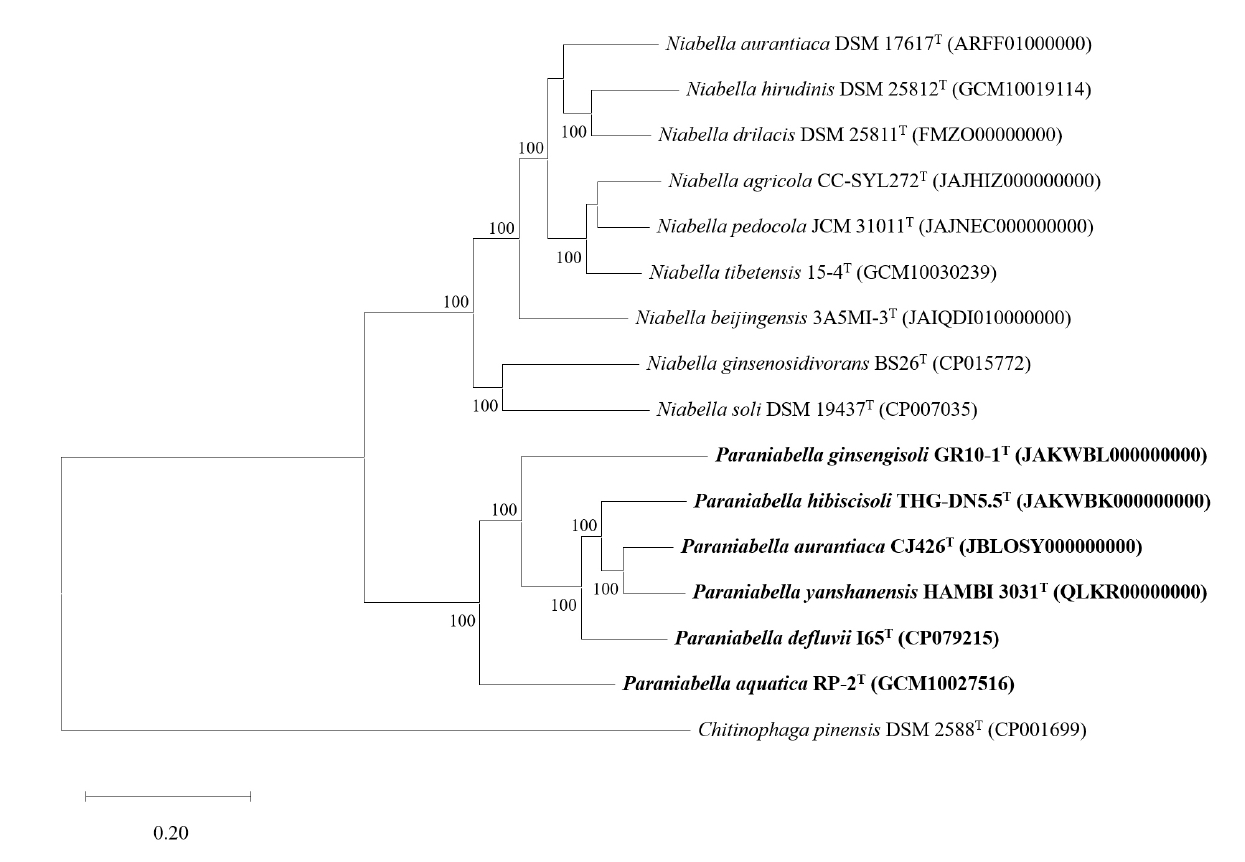
Fig. 4.Comparative genomic analysis of strain CJ426T and related type strains. (A) Pangenome analysis illustrates flower plots. The analysis shows the distribution of orthologous gene families calculated separately for 1) members of the genus Niabella, 2) members of the proposed genus Paraniabella, and 3) all strains from both genera combined. The numbers represent the counts of shared or unique orthologous gene families between the different strains. (B) Circular genome comparison of strain CJ426T and all related type strains based on whole-genome alignment. The reference genome is shown in the innermost circle, with concentric rings representing the genomic features of each strain. Red and blue colors indicated regions of similarity.
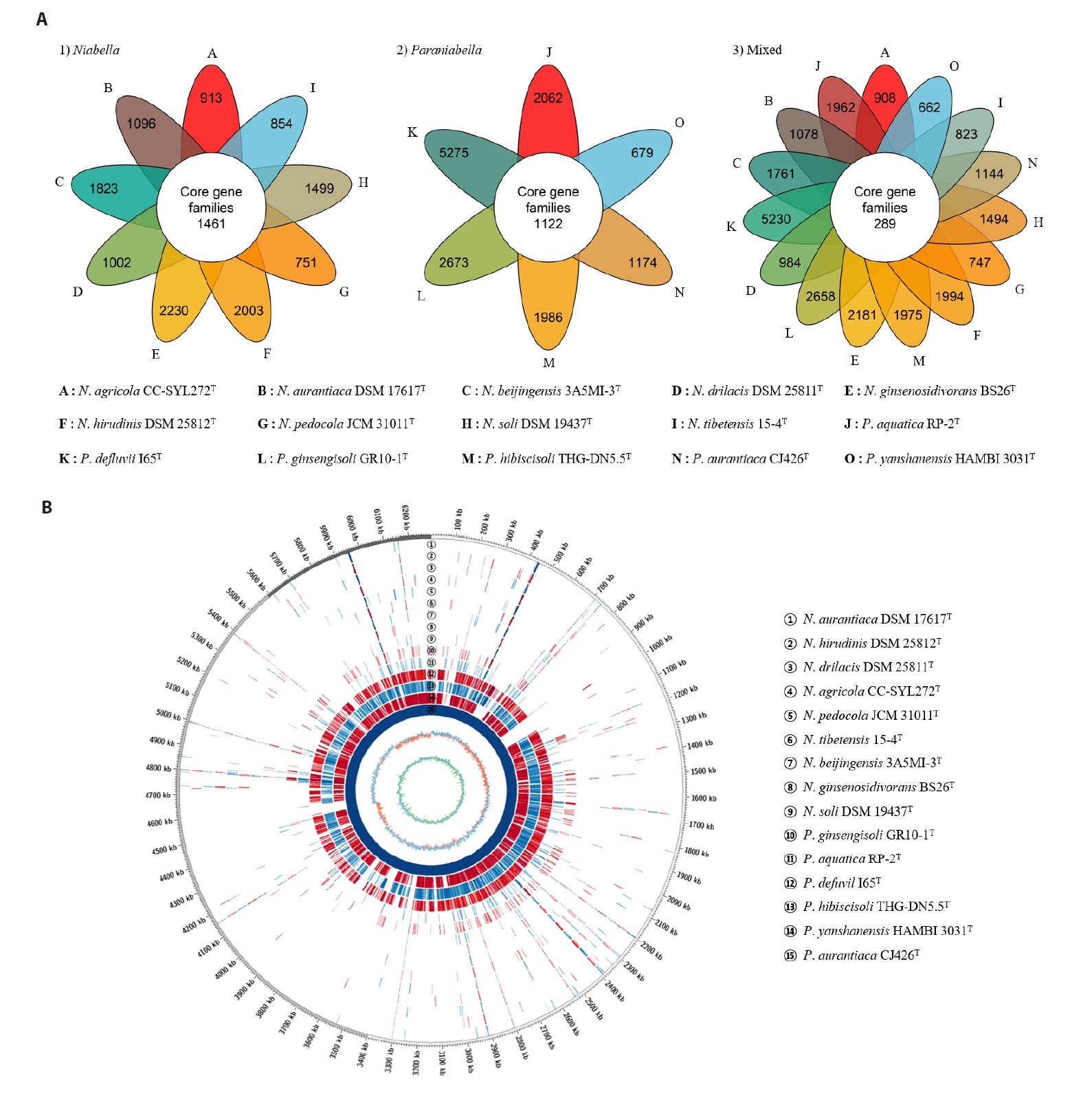
Table 1.Comparison of intra- and inter-group OGRI values
|
OGRI values (%) |
Group Ⅰ |
Group Ⅱ |
Group Ⅲ |
|
Group Ⅰ |
ANI |
73.6–84.6 |
|
|
|
AAI |
76.1–90.0 |
|
POCP |
55.0–78.7 |
|
Group Ⅱ |
ANI |
69.5–70.7 |
73.7–83.4 |
|
|
AAI |
67.5–69.3 |
75.8–88.4 |
|
POCP |
47.2–59.9 |
62.4–80.5 |
|
Group Ⅲ |
ANI |
70.0–71.2 |
74.2–75.5 |
NA |
|
AAI |
67.9–68.9 |
75.7–78.7 |
|
POCP |
48.2–56.7 |
58.4–66.3 |
Table 2.General genomic features of strain CJ426T and related type strains within the genera Niabella and Paraniabella
|
No |
Type strains |
Accession number |
Size (Mbp) |
GC contents (%) |
Number of |
|
contigs |
CDSs |
rRNA |
tRNA |
|
1 |
N. agricola CC-SYL272T
|
JAJHIZ000000000†
|
6.3 |
47.1 |
3 |
5,286 |
6 |
48 |
|
2 |
N. aurantiaca DSM 17617T
|
ARFF01000000†
|
5.9 |
48.6 |
25 |
4,870 |
12 |
50 |
|
3 |
N. beijingensis 3A5MI-3T
|
JAIQDI010000000†
|
6.6 |
47.1 |
3 |
5,604 |
3 |
43 |
|
4 |
N. drilacis DSM 25811T
|
FMZO00000000†
|
6.1 |
47.8 |
45 |
5,189 |
3 |
46 |
|
5 |
N. ginsenosidivorans BS26T
|
CP015772†
|
5.6 |
44.5 |
1 |
4,741 |
6 |
44 |
|
6 |
N. hirudinis DSM 25812T
|
GCM10019114‡
|
5.4 |
46.9 |
1 |
6,674 |
6 |
53 |
|
7 |
N. pedocola JCM 31011T
|
JAJNEC000000000†
|
6.5 |
47.3 |
11 |
5,310 |
3 |
42 |
|
8 |
N. soli DSM 19437T
|
CP007035†
|
4.7 |
45.2 |
1 |
3,888 |
6 |
41 |
|
9 |
N. tibetebsis 15-4T
|
GCM10030239‡
|
6.6 |
47.0 |
54 |
5,412 |
6 |
48 |
|
10 |
P. aquatica RP-2T
|
GCM10027516‡
|
5.4 |
42.0 |
40 |
4,539 |
3 |
38 |
|
11 |
P. aurantiaca CJ426T
|
JBLOSY000000000*
|
6.3 |
41.9 |
3 |
5,310 |
6 |
45 |
|
12 |
P. defluvii I65T
|
CP079215†
|
6.1 |
43.0 |
1 |
9,449 |
6 |
43 |
|
13 |
P. ginsengisoli GR10-1T
|
JAKWBL000000000†
|
4.5 |
38.5 |
5 |
5,443 |
6 |
44 |
|
14 |
P. hibiscisoli THG-DN5.5T
|
JAKWBK000000000†
|
6.1 |
42.1 |
5 |
8,185 |
6 |
42 |
|
15 |
P. yanshanensis HAMBI 3031T
|
QLKR00000000†
|
5.5 |
42.7 |
25 |
4,607 |
3 |
41 |
Table 3.Differential characteristics of strain CJ426T and related type strains of the family Chitinophagaceae. Strains: 1, CJ426T (this study); 2, N. aurantiaca DSM 17617T (Kim et al., 2007); 3, Terrimonas ferruginea DYT (Jin et al., 2013; Xie and Yokota, 2006); 4, Niastella hibisci THG-YS3.2.1T (Yan et al., 2016); 5, Sediminibacterium salmoneum NBRC 103935T (Kim et al., 2013; Qu and Yuan, 2008); 6, Niveitalea solisilvae 6-4T (Hyeon et al., 2017); 7, Flavihumibacter solisilvae 3-3T (Lee et al., 2014). Symbols: +, Positive; W, weakly positive; -, negative; ND, data not available. All strains are positive for activity of β-galactosidase, and esculin hydrolysis. All strains are negative for the assimilation of D-mannitol, potassium gluconate, capric acid, adipic acid, malic acid, trisodium citrate and phenylacetic acid.
|
Characteristic |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Isolation source†
|
Soil |
Soil |
Soil |
Soil |
Sediment |
Soil |
Soil |
|
Colony color‡
|
O |
O |
SR |
Y |
SP |
W |
Y |
|
Cell morphology |
Rod |
Rod |
Rod |
Rod |
Rod |
Rod |
Rod |
|
Cell motility |
- |
- |
- |
- |
+ |
- |
- |
|
Growth |
|
Temperature (°C) |
4–37 |
10–35 |
10–37 |
10–40 |
18–37 |
20–35 |
20–37 |
|
pH |
5–12 |
5–8 |
ND |
6–8 |
6.0–7.5 |
6.0–9.0 |
5.5–9.5 |
|
NaCl (%, w/v) |
0–2 |
0–3 |
0–1 |
0–1 |
0–1 |
0 |
0–0.5 |
|
Nirate reduction |
- |
- |
+ |
- |
- |
+ |
- |
|
Indole production |
+ |
+ |
- |
- |
- |
- |
- |
|
Fermentation of |
|
Glucose |
- |
- |
- |
- |
+ |
- |
- |
|
Hydrolysis of |
|
Casein |
+ |
+ |
- |
- |
- |
+ |
+ |
|
DNA |
- |
- |
+ |
- |
- |
ND |
ND |
|
Starch |
+ |
- |
+ |
- |
- |
- |
- |
|
CM-Cellulose |
+ |
+ |
- |
+ |
- |
ND |
ND |
|
Gelatin (API 20NE) |
+ |
- |
+ |
+ |
- |
- |
+ |
|
Enzyme of activity of: |
|
Catalase |
+ |
+ |
w |
+ |
+ |
- |
+ |
|
Oxidase |
- |
- |
+ |
- |
+ |
+ |
+ |
|
Urease |
+ |
- |
- |
- |
- |
- |
- |
|
Arginine dihydrolase |
+ |
- |
- |
- |
- |
- |
- |
|
Assimilation of: |
|
D-Glucose |
+ |
+ |
+ |
+ |
+ |
- |
+ |
|
L-Arabinose |
+ |
+ |
+ |
- |
- |
- |
+ |
|
D-Mannose |
+ |
+ |
w |
- |
+ |
- |
- |
|
N-Acetyl-glucosamine |
+ |
+ |
+ |
- |
- |
- |
+ |
|
D-Maltose |
+ |
+ |
+ |
- |
+ |
- |
+ |
|
G + C content (mol%) |
41.9*
|
45.0 |
48.9 |
45.3 |
40.6 |
45.8 |
49.5 |
|
Major polar lipids⁑
|
PE |
ND |
PE |
PME, PE |
PE |
PE |
PE |
Table 4.Fatty acid compositions (%) of strain CJ426T and related type strains of the family Chitinophagaceae. Strains: 1, CJ426T (this study); 2, N. aurantiaca DSM 17617T (Kim et al., 2007); 3, Terrimonas ferruginea DYT (Xie and Yokota, 2006); 4, Niastella hibisci THG-YS3.2.1T (Yan et al., 2016); 5, Sediminibacterium salmoneum NBRC 103935T (Qu and Yuan, 2008); 6, Niveitalea solisilvae 6-4T (Hyeon et al., 2017); 7, Flavihumibacter solisilvae 3-3T (Lee et al., 2014). Data are percentages of the total fatty acids (components < 1.0% in all strains are not shown). TR, trace amounts (< 1.0%); -, not detected; Major components (> 10.0%) are highlighted in bold type.
|
Fatty acid |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Straight |
|
C14:0
|
1.1 |
- |
TR |
TR |
- |
- |
- |
|
C15:0
|
- |
- |
2.7 |
- |
TR |
- |
- |
|
C16:0
|
5.9 |
3.5 |
1.7 |
8.3 |
1.2 |
2.6 |
1.7 |
|
C18:0
|
- |
- |
- |
3.7 |
- |
- |
- |
|
Branched |
|
anteiso-C12:0
|
- |
- |
- |
2.3 |
- |
- |
- |
|
iso-C13:0
|
- |
- |
TR |
- |
- |
- |
1.3 |
|
iso-C14:0
|
- |
- |
1.1 |
1.6 |
2.5 |
1.0 |
- |
|
iso-C15:0
|
38.2
|
33.7
|
28.4
|
14.3
|
17.5
|
34.0
|
39.8
|
|
iso-C15:1
|
- |
- |
26.2
|
- |
- |
- |
- |
|
anteiso-C15:0
|
- |
1.6 |
TR |
5.0 |
9.5 |
11.2
|
2.5 |
|
iso-C15:1 G |
20.2
|
22.3
|
- |
9.3 |
24.1
|
12.6
|
24.2
|
|
anteiso-C15:1 A |
- |
- |
- |
1.6 |
7.4 |
2.0 |
- |
|
iso-C16:0
|
- |
- |
1.4 |
3.5 |
1.1 |
2.4 |
- |
|
iso-C16:0 G |
- |
- |
- |
2.2 |
- |
- |
- |
|
Unsaturated |
|
C14:1 ω5c
|
- |
- |
- |
- |
- |
1.0 |
- |
|
C16:1 ω5c
|
1.4 |
- |
- |
- |
TR |
6.6 |
6.1 |
|
C17:1 ω6c
|
- |
- |
- |
3.3 |
- |
- |
- |
|
C18:3 ω6c (6,9,12) |
- |
- |
- |
2.5 |
- |
- |
- |
|
Hydroxy |
|
C15:0 2-OH |
- |
- |
TR |
1.1 |
1.9 |
- |
- |
|
iso-C15:0 3-OH |
2.8 |
2.9 |
2.2 |
- |
7.4 |
2.8 |
3.7 |
|
C16:0 3-OH |
2.0 |
2.4 |
2.5 |
1.5 |
1.8 |
1.6 |
- |
|
iso-C16:0 3-OH |
- |
- |
TR |
6.1 |
7.7 |
1.0 |
- |
|
C17:0 2-OH |
- |
- |
- |
2.3 |
2.2 |
1.2 |
- |
|
iso-C17:0 3-OH |
14.0
|
15.5
|
15.3
|
19.1
|
7.7 |
12.2
|
3.0 |
|
Summed feature 3†
|
7.2 |
10.6
|
11.2
|
11.1
|
TR |
4.4 |
11.1
|
References
- Baker GC, Smith JJ, Cowan DA. 2003. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods. 55: 541–555. ArticlePubMed
- Barco RA, Garrity GM, Scott JJ, Amend JP, Nealson KH, et al. 2020. A genus definition for Bacteria and Archaea based on a standard genome relatedness index. mBio. 11: e02475–19. ArticlePubMedPMCLink
- Bates RG, Bower VE. 1956. Alkaline solutions for pH control. Anal Chem. 28: 1322–1324. Article
- Blin K, Shaw S, Medema MH, Weber T. 2024. The antiSMASH database version 4: additional genomes and BGCs, new sequence-based searches and more. Nucleic Acids Res. 52: D586–D589. ArticlePubMedPDF
- Chalita M, Kim YO, Park S, Oh HS, Cho JH, et al. 2024. EzBioCloud: a genome-driven database and platform for microbiome identification and discovery. Int J Syst Evol Microbiol. 74: 006421.ArticlePubMedPMC
- Chan PP, Lin BY, Mak AJ, Lowe TM. 2021. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49: 9077–9096. ArticlePubMedPMCPDF
- Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, et al. 2018. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 68: 461–466. ArticlePubMed
- CLSI. 2025. Performance standards for antimicrobial susceptibility testing. 35 ed., vol. 45. Clinical and laboratory standards institute.
- Collins MD. 1985. 11 analysis of isoprenoid quinones. In Bergan T. (ed.), Methods in microbiology, vol. 18, pp. 329–366. Academic press. Article
- Cumsille A, Durán RE, Rodríguez-Delherbe A, Saona-Urmeneta V, Cámara B, et al. 2023. GenoVi, an open-source automated circular genome visualizer for bacteria and archaea. PLoS Comput Biol. 19: e1010998. ArticlePubMedPMC
- Dahal RH, Kim J. 2016. Niabella pedocola sp. nov., isolated from soil. Int J Syst Evol Microbiol. 66: 2650–2656. ArticlePubMed
- Dai J, Jiang F, Wang Y, Yu B, Qi H, et al. 2011. Niabella tibetensis sp. nov., isolated from soil, and emended description of the genus Niabella. Int J Syst Evol Microbiol. 61: 1201–1205. ArticlePubMed
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5: 113.ArticlePubMedPMCPDF
- Fan G, Sun Q, Sun Y, Liu D, Li S, et al. 2024. GCM and gcType in 2024: comprehensive resources for microbial strains and genomic data. Nucleic Acids Res. 53: D763–D771. ArticlePMCPDF
- Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, et al. 2021. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep. 11: 12728.ArticlePubMedPMCPDF
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 39: 783–791. ArticlePubMedLink
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39: W29–W37. ArticlePubMedPMC
- Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, et al. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 74: 2461–2470. ArticlePubMedPMCLink
- Glaeser SP, Galatis H, Martin K, Kämpfer P. 2013. Niabella hirudinis and Niabella drilacis sp. nov., isolated from the medicinal leech Hirudo verbana. Int J Syst Evol Microbiol. 63: 3487–3493. ArticlePubMed
- Guo SZ, Wu T, Zhu HZ, Yan L, Liu ZP, et al. 2022. Niabella beijingensis sp. nov. and Thermomonas beijingensis sp. nov., two bacteria from constructed wetland. Int J Syst Evol Microbiol. 72: 005280.ArticlePubMedPMC
- Hunt M, De Silva N, Otto TD, Parkhill J, Keane JA, et al. 2015. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16: 294.ArticlePubMedPMCPDF
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 11: 119.ArticlePubMedPMCPDF
- Hyeon JW, Lee HJ, Jeong SE, Cho GY, Jeon CO. 2017. Niveitalea solisilvae gen. nov., sp. nov., isolated from forest soil and emended description of the genus Flavihumibacter Zhang et al. 2010. Int J Syst Evol Microbiol. 67: 1374–1380. ArticlePubMed
- Jin D, Wang P, Bai Z, Jin B, Yu Z, et al. 2013. Terrimonas pekingensis sp. nov., isolated from bulking sludge, and emended descriptions of the genus Terrimonas, Terrimonas ferruginea, Terrimonas lutea and Terrimonas aquatica. Int J Syst Evol Microbiol. 63: 1658–1664. ArticlePubMed
- Jukes TH, Cantor CR. 1969. Chapter 24 - Evolution of protein molecules. In Munro HN. (ed.), Mammalian protein metabolism, pp. 21–132. Academic press.
- Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, et al. 2021. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49: D192–D200. ArticlePubMedPDF
- Kämpfer P, Lodders N, Falsen E. 2011. Hydrotalea flava gen. nov., sp. nov., a new member of the phylum Bacteroidetes and allocation of the genera Chitinophaga, Sediminibacterium, Lacibacter, Flavihumibacter, Flavisolibacter, Niabella, Niastella, Segetibacter, Parasegetibacter, Terrimonas, Ferruginibacter, Filimonas and Hydrotalea to the family Chitinophagaceae fam. nov. Int J Syst Evol Microbiol. 61: 518–523. ArticlePubMed
- Kim J, Na SI, Kim D, Chun J. 2021a. UBCG2: Up-to-date bacterial core genes and pipeline for phylogenomic analysis. J Microbiol. 59: 609–615. ArticlePDF
- Kim YJ, Nguyen NL, Weon HY, Yang DC. 2013. Sediminibacterium ginsengisoli sp. nov., isolated from soil of a ginseng field, and emended descriptions of the genus Sediminibacterium and of Sediminibacterium salmoneum. Int J Syst Evol Microbiol. 63: 905–912. ArticlePubMed
- Kim M, Oh HS, Park SC, Chun J. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 64: 346–351. ArticlePubMed
- Kim D, Park S, Chun J. 2021b. Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J Microbiol. 59: 476–480. ArticlePDF
- Kim BY, Weon HY, Yoo SH, Hong SB, Kwon SW, et al. 2007. Niabella aurantiaca gen. nov., sp. nov., isolated from a greenhouse soil in Korea. Int J Syst Evol Microbiol. 57: 538–541. ArticlePubMed
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, et al. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19: 1639–1645. ArticlePubMedPMC
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5: R12.ArticlePubMedPMCPDF
- Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32: 11–16. ArticlePubMedPMC
- Lee HJ, Jeong SE, Cho MS, Kim S, Lee SS, et al. 2014. Flavihumibacter solisilvae sp. nov., isolated from forest soil. Int J Syst Evol Microbiol. 64: 2897–2901. ArticlePubMed
- Lee I, Kim YO, Park SC, Chun J. 2016. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 66: 1100–1103. ArticlePubMed
- Lin SY, Tsai CF, Hameed A, Lee TH, Young CC. 2022. Niabella agricola sp. nov., isolated from paddy soil. Int J Syst Evol Microbiol. 72: 005559.Article
- Luo C, Rodriguez-R LM, Konstantinidis KT. 2014. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 42: e73. ArticlePubMedPMC
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. BUSCO update: novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol. 38: 4647–4654. ArticlePubMedPMCPDF
- Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 14: 60.ArticlePubMedPMCPDF
- Meier-Kolthoff JP, Klenk HP, Goker M. 2014. Taxonomic use of DNA G + C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 64: 352–356. ArticlePubMed
- Meier-Kolthoff JP, Sardà Carbasse J, Peinado-Olarte RL, Göker M. 2022. Peinado-Olarte RL, Göker M. 2022. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50: D801–D807. ArticlePubMed
- Min J, Son Y, Park Y, Park W. 2024. Niabella defluvii sp. nov., isolated from influent water of a wastewater treatment plant. Int J Syst Evol Microbiol. 74: 006425.Article
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, et al. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 37: 1530–1534. ArticlePubMedPMCPDF
- Minnikin DE, O'Donnell AG, Goodfellow M, Alderson G, Athalye M, et al. 1984. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Methods. 2: 233–241. Article
- Nawrocki EP, Eddy SR. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29: 2933–2935. ArticlePubMedPMCPDF
- Ngo HTT, Trinh H, Yan ZF, Moya G, Kook M, et al. 2017. Niabella hibiscisoli sp. nov., isolated from soil of a Rose of Sharon garden. Int J Syst Evol Microbiol. 67: 784–788. ArticlePubMed
- O'Leary NA, Cox E, Holmes JB, Anderson WR, Falk R, et al. 2024. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 11: 732.ArticlePubMedPMC
- Parte AC, Sardà Carbasse J, Meier-Kolthoff JP, Reimer LC, Göker M. 2020. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol. 70: 5607–5612. ArticlePubMedPMC
- Philippon A, Slama P, Dény P, Labia R. 2016. A structure-based classification of class A β-lactamases, a broadly diverse family of enzymes. Clin Microbiol Rev. 29: 29–57. ArticlePubMedLink
- Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, et al. 2014. A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol. 196: 2210–2215. ArticlePubMedPMCLink
- Qu JH, Yuan HL. 2008. Sediminibacterium salmoneum gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from sediment of a eutrophic reservoir. Int J Syst Evol Microbiol. 58: 2191–2194. ArticlePubMed
- Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, et al. 2017. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46: D624–D632. ArticlePMC
- Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 106: 19126–19131. ArticlePubMedPMC
- Riesco R, Trujillo ME. 2024. Update on the proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 74: 006300.ArticlePubMedPMC
- Robertson J, Nash JHE. 2018. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom. 4: e000206. ArticlePubMedPMC
- Sasser M. 1990. Identification of bacteria by gas chromatography of cellular fatty acids. MIDI technical note 101. MIDI, Inc., USA.
- Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, et al. 2021. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom. 7: 000685.ArticlePubMedPMC
- Siddiqi MZ, Im WT. 2016. Niabella aquatica sp. nov., isolated from lake water. Int J Syst Evol Microbiol. 66: 2774–2779. ArticlePubMed
- Tamura K, Stecher G, Kumar S. 2021. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 38: 3022–3027. ArticlePubMedPMCPDF
- Tindall BJ. 1990a. A comparative study of the lipid composition of Halobacterium saccharovorum from various sources. Syst Appl Microbiol. 13: 128–130. Article
- Tindall BJ. 1990b. Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol Lett. 66: 199–202. Article
- Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, et al. 2020. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21: 180.ArticlePubMedPMCPDF
- UniProt C. 2021. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49: D480–D489. ArticlePubMed
- Wang H, Zhang YZ, Man CX, Chen WF, Sui XH, et al. 2009. Niabella yanshanensis sp. nov., isolated from the soybean rhizosphere. Int J Syst Evol Microbiol. 59: 2854–2856. ArticlePubMed
- Weon HY, Kim BY, Joa JH, Kwon SW, Kim WG, et al. 2008. Niabella soli sp. nov., isolated from soil from Jeju Island, Korea. Int J Syst Evol Microbiol. 58: 467–469. ArticlePubMed
- Weon HY, Yoo SH, Kim BY, Son JA, Kim YJ, et al. 2009. Niabella ginsengisoli sp. nov., isolated from soil cultivated with Korean ginseng. Int J Syst Evol Microbiol. 59: 1282–1285. ArticlePubMed
- Xie CH, Yokota A. 2006. Reclassification of [Flavobacterium] ferrugineum as Terrimonas ferruginea gen. nov., comb. nov., and description of Terrimonas lutea sp. nov., isolated from soil. Int J Syst Evol Microbiol. 56: 1117–1121. ArticlePubMed
- Yan ZF, Lin P, Wang YS, Gao W, Li CT, et al. 2016. Niastella hibisci sp. nov., isolated from rhizosphere soil of mugunghwa, the Korean national flower. Int J Syst Evol Microbiol. 66: 5218–5222. ArticlePubMed
- Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, et al. 2014. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 12: 635–645. ArticlePubMedPDF
- Yi KJ, Im WT, Kim DW, Liu QM, Kim SK. 2015. Niabella ginsenosidivorans sp. nov., isolated from compost. J Microbiol. 53: 762–766. ArticlePubMedPDF
- Yuan C, Liu B, Wang L, Long W, Ke Z, et al. 2023. Paraflavisolibacter caeni gen. nov., sp. nov., a novel taxon within the family Chitinophagaceae isolated from sludge. Int J Syst Evol Microbiol. 73: 005849.Article
- Zheng J, Ge Q, Yan Y, Zhang X, Huang L, et al. 2023. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51: W115–W121. ArticlePubMedPMCPDF
- Zhou Z, Tran PQ, Breister AM, Liu Y, Kieft K, et al. 2022. METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome. 10: 33.ArticlePubMedPMCPDF
Citations
Citations to this article as recorded by

- Validation List no. 229: valid publication of new names and new combinations effectively published outside the IJSEM
Aharon Oren, Markus Göker
International Journal of Systematic and Evolutionary Microbiology
.2026;[Epub] CrossRef - Notification of changes in taxonomic opinion previously published outside the IJSEM. List of Changes in Taxonomic Opinion no. 44
Aharon Oren, Markus Göker
International Journal of Systematic and Evolutionary Microbiology
.2026;[Epub] CrossRef


 ePub Link
ePub Link Cite this Article
Cite this Article