ABSTRACT
- Three Gram-stain-negative, strictly aerobic, motile bacterial strains, designated IMCC44359T, IMCC44632T, and IMCC44653, were isolated from coastal surface seawater collected near Jangbong Island in the Yellow Sea. Phylogenetic analyses based on 16S rRNA gene and whole-genome sequences assigned the isolates to the genus Aliikangiella. Strains IMCC44632T and IMCC44653 shared identical 16S rRNA gene sequences and exhibited high genomic relatedness (99.0% average nucleotide identity and 92.0% digital DNA-DNA hybridization), indicating that they represent a single species. In contrast, strain IMCC44359T showed low genomic relatedness to these strains and to previously validly published Aliikangiella species, supporting its recognition as a distinct species. The genome of IMCC44359T (5.95 Mbp; 36.5 mol% G + C) is substantially larger than those of IMCC44632T and IMCC44653 (~3.75 Mbp; 40.1–40.2 mol% G + C), and all genomes encode aerobic chemoorganotrophic metabolism and biochemical capacities consistent with adaptation to marine environments. The isolates grew under mesophilic and moderately halophilic conditions typical of coastal seawater bacteria, with growth occurring at ranges at 10–40℃, pH 6.0–9.0, and 0.5–7.5% NaCl (optimum, 30℃, pH 7.0–8.0, and 2.0–3.0% NaCl). All strains contained ubiquinone-8 (Q-8) as the sole respiratory quinone, and phosphatidylethanolamine, phosphatidylglycerol, and diphosphatidylglycerol were the major polar lipids. The dominant cellular fatty acids were iso-C15:0 and summed feature 9 (iso-C17:1 ω9c and/or C16:0 10-methyl). Integrated phylogenetic, genomic, phenotypic evidence supported the recognition of two novel species within the genus Aliikangiella, for which the names Aliikangiella litoralis sp. nov. (type strain IMCC44359T = KCTC 18089T = JCM 37879T = HNIBRBA19635T) and Aliikangiella aequoris sp. nov. (type strain IMCC44632T = KCTC 18090T = JCM 37880T = HNIBRBA19636T) are proposed.
-
Keywords: Aliikangiella litoralis, Aliikangiella aequoris, polyphasic taxonomy, marine bacteria, novel species, genome
Introduction
The family Kangiellaceae currently includes three genera, Kangiella, Pleionea, and Aliikangiella. The type genus Kangiella (Yoon et al., 2004) encompasses 11 validly published species that are non-motile rods, whereas Pleionea and Aliikangiella include both non-motile and flagellated taxa, indicating phenotypic diversification within the family. Although Wang et al. (2020) proposed the family Pleioneaceae to accommodate Pleionea and Aliikangiella, this family is treated as a heterotypic synonym of Kangiellaceae in the LPSN (List of Prokaryotic names with Standing in Nomenclature) framework (Parte et al., 2020). Overall, members of these genera share broadly conserved chemotaxonomic features yet differ in motility and genome characteristics, consistent with diversification within the family Kangiellaceae.
The genus Aliikangiella (family Kangiellaceae, order Oceanospirillales) was established by Wang et al. (2015) with Aliikangiella marina as the type species, which was isolated from a culture of the marine microalga Picochlorum sp. The taxonomic scope of the genus was later expanded by the description of Aliikangiella coralliicola from coral-associated samples (Wang et al., 2020). At present, the genus comprises two validly published species and one not-yet-validly published species, “Aliikangiella maris” (Li et al., 2025), which was isolated from the marine alga Phaeocystis globosa. Notably, Aliikangiella has so far been recovered only from marine settings (e.g., microalgal culture systems and coral-associated habitats), implying adaptation to marine-associated ecological niches.
Species of Aliikangiella are typically mesophilic and moderately halophilic, with growth optima compatible with marine conditions. Reported chemotaxonomic traits include ubiquinone-8 (Q-8) as the major respiratory quinone and phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and diphosphatidylglycerol (DPG) as major polar lipids. Comparative genomics has also suggested that Aliikangiella possesses relatively large genomes (5.34–7.04 Mbp) compared with other Kangiellaceae genera such as Kangiella (2.46–2.85 Mbp) and Pleionea (4.04–5.19 Mbp), potentially reflecting broader functional capacity.
Island-associated coastal waters around the Korean Peninsula are biologically dynamic and have repeatedly yielded novel bacterial taxa (Han et al., 2025; Oh et al., 2026; Tak et al., 2024; Yang et al., 2024). During ongoing cultivation-based surveys of coastal microbial diversity, strains IMCC44359T, IMCC44632T, and IMCC44653 were isolated from surface seawater collected near Jangbong Island in the Yellow Sea. A polyphasic taxonomic assessment indicated that these isolates represent two previously unrecognized novel species within the genus Aliikangiella. In the present study, we report their phylogenetic, genomic, and phenotypic characteristics and propose Aliikangiella litoralis sp. nov. and Aliikangiella aequoris sp. nov.
Materials and Methods
Isolation and culture conditions
Strain IMCC44359T was isolated from coastal surface seawater collected at Jangbong Island, Incheon, Republic of Korea (37.55172° N, 126.33675° E) in June 2023. Strains IMCC44632T and IMCC44653 were obtained from seawater collected at a different coastal beach on Jangbong Island (37.54072° N, 126.32931° E) during the same period. Serially diluted seawater samples were spread on marine agar 2216 (MA; BD Difco, USA). After incubation at 20°C for 5 days, colonies were purified by three successive transfers. The isolates were routinely cultivated on MA at 30°C and preserved at −80°C as glycerol stocks (10%, v/v). For comparative phenotypic analyses, A. marina KCTC 42667T (= GYP-15T) and A. coralliicola KCTC 72442T (= M105T) were obtained from the Korean Collection for Type Cultures (KCTC) and grown on MA at 30°C for 5 day; meanwhile, the phenotypic characteristics of “A. maris” GXAS 306T were obtained from the published literature (Li et al., 2025), having been generated under growth conditions identical or comparable to those used in this study (e.g., MA at 30°C).
16S rRNA gene-based phylogenetic analysis
The 16S rRNA gene sequences of strains IMCC44359T, IMCC44632T, and IMCC44653 were obtained by PCR from cell lysates prepared in Tris–EDTA buffer using mechanical disruption with glass beads and repeated freeze–thaw cycles. Amplification was performed with universal bacterial primers 27F and 1492R (Weisburg et al., 1991). Sanger sequencing (Macrogen Inc., Korea) used primers 27F, 518F, 800R, and 1492R. Sequence comparisons were conducted using BLASTn against GenBank, and pairwise similarities were calculated with EzBioCloud (Chalita et al., 2024).
For phylogenetic reconstruction, sequences were aligned with the SINA aligner (SILVA Incremental Aligner; v1.2.12) (Pruesse et al., 2012) and curated in ARB (Ludwig et al., 2004). Tree inference was performed in MEGA X (Kumar et al., 2018) using maximum-likelihood (ML) (Felsenstein, 1981), neighbor-joining (NJ) (Saitou and Nei, 1987), and minimum-evolution (ME) (Rzhetsky and Nei, 1992) algorithms. For ML analysis, the best-fit nucleotide substitution model was determined in MEGA X based on the Bayesian Information Criterion (BIC), and the ML tree was reconstructed under the selected model. NJ and ME trees were generated using evolutionary distances calculated with the Kimura two-parameter model (Kimura, 1980). The robustness of tree topologies was evaluated by bootstrap analysis with 1,000 replicates (Felsenstein, 1985).
Whole-genome sequencing and analysis
Approximately 50 mg (wet weight) of biomass from MA plates was resuspended in DNA/RNA Shield (Zymo Research, USA) and shipped to MicrobesNG (UK). Whole-genome sequencing was performed using a hybrid approach that combined Illumina short-read and Oxford Nanopore long-read platforms. Illumina libraries were prepared with the Nextera XT DNA Library Preparation Kit and sequenced on a NextSeq 2000 platform to generate 250-bp paired-end reads. Nanopore libraries were prepared with the SQK-RBK114.96 kit and sequenced on a PromethION system with a FLO-PRO114M flow cell. Hybrid assemblies were produced using Hybracter v0.11.2 (Bouras et al., 2024). Genome completeness and contamination were evaluated with CheckM2 v1.1.0 (Chklovski et al., 2023).
Genome-based taxonomic analyses were conducted following the framework proposed by Riesco and Trujillo (2024). Species-level relatedness was assessed by calculating average nucleotide identity (ANI) values using JSpeciesWS (Richter et al., 2016) and by estimating digital DNA–DNA hybridization (dDDH) values with the Genome-to-Genome Distance Calculator (GGDC 3.0) (Meier-Kolthoff et al., 2013). For genus-level comparisons, average amino acid identity (AAI) values were calculated using EzAAI v1.2.3 (Kim et al., 2021), and the percentage of conserved proteins (POCP) was determined using the Nextflow-based POCP pipeline (hoelzer/pocp, v2.3.6) following Hölzer (2024), with protein alignments performed using DIAMOND. Reference genome sequences for comparative analyses were retrieved from the NCBI Datasets database (O’Leary et al., 2024).
Genome-based phylogenetic relationships were inferred using GTDB-Tk v2.4.0 (Chaumeil et al., 2022) with the bacterial bac120 marker gene set from the Genome Taxonomy Database (GTDB; release R226). The concatenated alignment of 120 single-copy marker genes was analyzed under the maximum-likelihood framework using RAxML v8.2.12 (Stamatakis, 2014). Functional genome annotation was performed with Prokka v1.14.6 (Seemann, 2014). Predicted protein sequences were assigned to KEGG orthologs using BlastKOALA (Kanehisa et al., 2016), and metabolic pathway completeness was assessed using the kegg-pathway-completeness tool v1.3.0 (https://github.com/EBI-Metagenomics/kegg-pathways-completeness-tool).
Physiological and biochemical characterization
Cell morphology was examined by transmission electron microscopy (CM200, Philips, Netherlands) after negative staining. Cells were suspended in 0.2 M cacodylate buffer, stained with 1% (w/v) uranyl acetate (Electron Microscopy Sciences, USA), and mounted on Formvar-coated copper grids (FCF300-CU; Ted Pella, USA). The Gram reaction was assessed using the KOH-based non-staining method (Powers, 1995). Catalase activity was tested with 3% (v/v) H2O2, and oxidase activity was determined using 1% (w/v) Kovac’s reagent (bioMérieux, France). Motility was assessed in soft MA (0.5%, BD Difco, USA) by observing outward diffusion from the inoculation point. Temperature-dependent growth was examined on MA at 4°C and at 10–50°C (5°C intervals). NaCl tolerance was tested on NaCl-free MA supplemented to final concentrations of 0–4.0% (w/v; 0.5% increments), 5%, 7.5%, 10%, 15%, and 20%. pH range and optimum were assessed in marine broth (MB; BD Difco, USA) adjusted to pH 3.5–10.5 (0.5-unit increments) using citrate, MES, MOPS, HEPES, Tris, and CHES buffers (Sigma-Aldrich, USA), with growth monitored by OD600 (UV2600; Shimadzu, Japan). Anaerobic growth was examined using the GasPakTM EZ Anaerobe Pouch System with Indicator (BD Diagnostics, USA). To verify anaerobic conditions, the anaerobic bacterium Ferrimonas sediminicola IMCC35001T was inoculated as a positive control. Hydrolytic activities were tested on MA supplemented with starch (1%, w/v), carboxymethyl cellulose (1%, w/v), chitin (1%, w/v), casein (3% skim milk, w/v), Tween 20 (1%, v/v), and or Tween 80 (1%, v/v) (Sigma-Aldrich, USA). DNA degradation was assessed using DNase Test Agar (BD Diagnostics, USA), and H2S production was tested on Triple Sugar Iron (TSI) agar (BD Difco, USA). Esculin hydrolysis activity was determined using Esculin Agar (MB-E2166, MB Cell). Additional biochemical characteristics were determined using API 20NE, API ZYM kits (bioMérieux, France), and the GEN III MicroPlate (Biolog, USA) with salinity adjusted to 2% NaCl.
Chemotaxonomic characterization
Fatty acid methyl ester (FAME) analysis was performed using biomass from IMCC44359T, IMCC44632T, IMCC44653, A. marina KCTC 42667T, and A. coralliicola KCTC 72442T grown on MA at 30°C for 5 days. Fatty acids were analyzed using an Agilent 7890 GC (Agilent Technologies, USA) with the Sherlock MIS (MIDI, USA) v6.1 and TSBA6 database (Sasser, 1990). Polar lipids were extracted as described by Minnikin et al. (1984) and separated by two-dimensional TLC on silica gel 60 F254 plates (Merck, Germany). Solvent systems were chloroform/methanol/water (60:30:4, v/v) in the first dimension and chloroform/acetic acid/methanol/water (40:7.5:6:1.8, v/v) in the second. Total lipids were visualized with molybdophosphoric acid, and lipid classes were detected using ninhydrin (aminolipids), molybdenum blue (phospholipids), α-naphthol (glycolipids), and Dragendorff’s reagent (phosphatidylcholine). Respiratory quinones were co-extracted and analyzed by reverse-phase HPTLC on RP-18 F254 plates (Merck, Germany) following Collins and Jones (1981).
Nucleotide sequence accession numbers
The 16S rRNA gene sequences of strains IMCC44359T, IMCC44632T, and IMCC44653 have been deposited in the GenBank/EMBL/DDBJ databases under accession numbers PX239437, PX239438, and PX239439, respectively. The complete genome sequences are available under accession numbers CP199292 (IMCC44359T), JBSKFV000000000 (IMCC44632T), and JBSKFW000000000 (IMCC44653).
Results and Discussion
16S rRNA gene-based phylogeny
Nearly full-length 16S rRNA gene sequences were obtained for IMCC44359T (1,464 bp), IMCC44632T (1,441 bp), and IMCC44653 (1,437 bp). Strains IMCC44359T and IMCC44632T shared 97.2% sequence similarity, which is below the commonly accepted 98.7% threshold for species delineation based on 16S rRNA gene similarity (Riesco and Trujillo, 2024), indicating that they represent distinct species-level lineages. In contrast, strains IMCC44632T and IMCC44653 exhibited identical 16S rRNA gene sequences (100% similarity), indicating that they may belong to the same species. Accordingly, genome-based relatedness analyses were performed to further clarify species delineation.
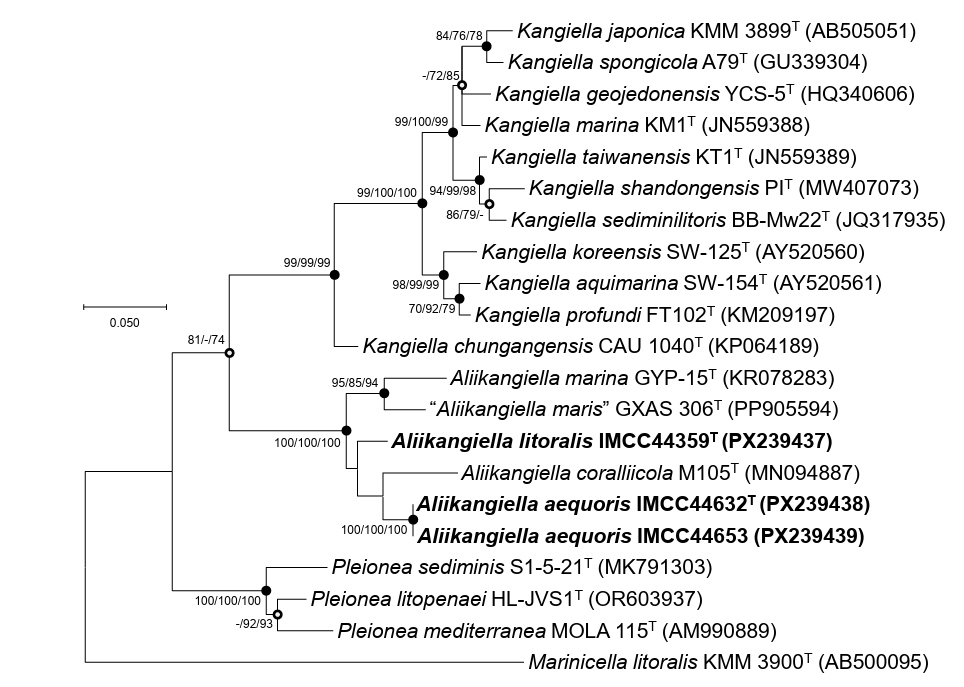
BLASTn/EzBioCloud comparisons and phylogenetic reconstructions supported assignment of the three strains to the genus Aliikangiella. Strain IMCC44359T exhibited the highest sequence similarity to “A. maris” GXAS 306T (96.0%), followed by A. marina GYP-15T (95.3%) and A. coralliicola M105T (95.3%). Both strains IMCC44632T and IMCC44653 exhibited 96.4%, 94.9%, and 94.5% similarity to A. coralliicola M105T, “A. maris” GXAS 306T, and A. marina GYP-15T, respectively. All similarity values were well below 98.7%, indicating that the IMCC isolates represent phylogenetically distinct lineages from previously described Aliikangiella species. In the 16S rRNA gene tree, strains IMCC44359T, IMCC44632T, and IMCC44653 clustered within the family Kangiellaceae and formed a distinct, strongly supported clade within the genus Aliikangiella, positioned close to the genus Pleionea (Fig. 1).
Genome characteristics and phylogenomic analysis
Each genome was assembled into a single circular chromosome. Genome sizes were 5,951,045 bp (IMCC44359T), 3,753,390 bp (IMCC44632T), and 3,750,766 bp (IMCC44653), with DNA G + C contents of 36.5%, 40.1%, and 40.2%, respectively. CheckM2 estimates indicated high completeness (100% for all) with low contamination (1.7% for IMCC44359T; < 0.5% for IMCC44632T and IMCC44653). Genome annotation predicted 4,928 protein-coding genes for IMCC44359T and 3,229/3,231 for IMCC44632T/IMCC44653. Strain IMCC44359T contained 50 tRNA genes and six rRNA genes, whereas IMCC44632T and IMCC44653 each harbored 43 tRNA genes and nine rRNA genes. The substantial genome size difference between strain IMCC44359T (~5.95 Mb) and strains IMCC44632T and IMCC44653 (~3.75 Mb) is unlikely to be attributable to assembly artifacts or contamination, given the high completeness and low contamination of the genome assemblies. This difference likely reflects variation in gene content and functional potential between the Aliikangiella species, consistent with their genomic distinctiveness. Comparative genome features for the three strains and three Aliikangiella species are summarized in Table S1. Overall genome relatedness indices clearly supported the presence of two species among the isolates. Strains IMCC44632T and IMCC44653 exhibited ANI and dDDH values of 99.0% and 92.0%, respectively, consistent with a single genomic species. In contrast, comparisons between IMCC44359T and IMCC44632T/IMCC44653 yielded ANI and dDDH values of 69.9% and 18.6%, respectively. Relative to the three Aliikangiella species, all three strains showed low genomic relatedness values (ANI 68.6–71.3%; dDDH 18.0–20.7%). Using the accepted thresholds of 95–96% ANI and 70% dDDH (Riesco and Trujillo, 2024), these data indicate that IMCC44359T represents one novel species and that IMCC44632T/IMCC44653 together represent a second novel species within the genus Aliikangiella. Genus-level affiliation was further supported by AAI values of 68.1–72.0% (Konstantinidis and Tiedje, 2007) and POCP values of 53.3–63.9% (Qin et al., 2014) relative to Aliikangiella species (Table S3). Concordantly, phylogenomic inference placed the three strains in a distinct, well-supported clade within the genus Aliikangiella (Fig. 2).
Genome-based metabolic reconstruction using KEGG module profiling revealed that the three strains share a heterotrophic, aerobic metabolic lifestyle (Table S4). In all genomes, the upper portion of the Embden–Meyerhof–Parnas pathway for glucose utilization (KEGG module ID: M00001) was incomplete, whereas the downstream glycolytic module for three-carbon intermediates (M00002) was complete. All strains encoded the non-oxidative pentose phosphate pathway (M00007) and PRPP biosynthesis (M00005) but lacked the oxidative pentose phosphate module (M00006), a configuration observed in other marine heterotrophic bacteria such as SAR11 and SAR86 (Dupont et al., 2012; Giovannoni et al., 2005). The Entner–Doudoroff pathway (M00008) was present only in IMCC44359T and absent from IMCC44632T and IMCC44653, indicating strain-level variation in carbohydrate catabolism. All three genomes encoded a complete TCA cycle (M00010 and M00011) and pyruvate oxidation (M00307), consistent with aerobic respiration. No canonical carbon fixation pathways (Calvin–Benson–Bassham cycle, reductive TCA cycle, Wood–Ljungdahl pathway, or 3-hydroxypropionate cycles) were detected, providing no genomic evidence for autotrophy or mixotrophy. Oxidative phosphorylation modules were complete in all strains, including NADH dehydrogenase (M00144), succinate dehydrogenase (M00149), cytochrome bc1 complex (M00151), cytochrome c oxidase (M00155), and F-type ATP synthase (M00157). Fatty acid biosynthesis and β-oxidation pathways were also considered complete (M00082, M00083, M00086, M00087). In addition, broad cofactor and vitamin biosynthesis potential was inferred (e.g., NAD, ubiquinone, coenzyme A, heme, biotin, riboflavin, siroheme, molybdenum cofactor, lipoic acid, and cobalamin). Collectively, these features are consistent with aerobic chemoorganotrophic marine bacteria that conserve energy via respiratory oxidation of organic substrates, in line with previously described members of Aliikangiella.
Phenotypic characteristics
Transmission electron microscopy demonstrated that cells of all three strains were rod-shaped and motile by a single polar flagellum (Fig. S1), in agreement with the presence of flagellar assembly genes identified in their genomes. Cell dimensions were approximately 0.3–0.5 × 1.7–2.7 µm for IMCC44359T, 0.4–0.8 × 1.4–2.6 µm for IMCC44632T, and 0.6–0.8 × 3.1–3.8 µm for IMCC44653. The physiological and biochemical properties of strains IMCC44359T, IMCC44632T, and IMCC44653 are summarized in Tables 1 and S2, and the species protologues. The three IMCC isolates could be differentiated from previously described Aliikangiella species by distinct patterns of NaCl tolerance, starch hydrolysis, trypsin activity, and substrate oxidation profiles, supporting phenotypic separation.
Cellular fatty acid compositions of strains IMCC44359T, IMCC44632T, and IMCC44653, along with those of the type strains of the validly published Aliikangiella species, are presented in Table 2. Overall, the fatty acid profiles were consistent with affiliation to the genus Aliikangiella, being dominated by iso-C15:0 and summed feature 9 (iso-C17:1 ω9c and/or C16:0 10-methyl). The dominance of iso-C15:0 and summed feature 9 has also been reported for “A. maris” GXAS 306T (Li et al., 2025). The three novel strains shared several major fatty acids present at proportions greater than 10%, including iso-C15:0 (27.6%, 42.7%, and 42.9% in IMCC44359T, IMCC44632T, and IMCC44653, respectively) and summed feature 9 (19.2%, 26.9%, and 27.4%, respectively). Despite this overall similarity, clear quantitative differences were observed, particularly in the proportions of C14:0, C16:0, and iso-C11:0 3-OH. These quantitative differences, together with variation in several minor unsaturated fatty acids, contributed to chemotaxonomic differentiation among the three strains and from previously described members of the genus.
All three strains possessed Q-8 as the sole respiratory quinone, consistent with the established chemotaxonomic characteristics of the genus Aliikangiella. The polar lipid profile of IMCC44359T consisted of phosphatidylethanolamine (PE), phosphatidylglycerol (PG), diphosphatidylglycerol (DPG), one unidentified aminolipid (AL), and one unidentified lipid (Fig. S2). Strains IMCC44632T and IMCC44653 exhibited comparable profiles comprising PE, PG, DPG, one unidentified aminophospholipid (APL), and three unidentified lipids. The shared presence of PE, PG, and DPG as major polar lipids corresponds with previously reported lipid patterns for members of the genus. Collectively, the fatty acid composition, quinone, and polar lipid characteristics provide coherent chemotaxonomic evidence supporting placement of these strains within the genus Aliikangiella.
Taxonomic conclusion
Integrated evidence from 16S rRNA gene phylogeny, genome-scale relatedness indices, and physiological and chemotaxonomic characterization supported the placement of strains IMCC44359T, IMCC44632T, and IMCC44653 within the genus Aliikangiella. Strains IMCC44632T and IMCC44653 exhibited genome relatedness values consistent with membership in a single species, whereas IMCC44359T was clearly separated from these strains and from the two validly published Aliikangiella species by ANI and dDDH values well below the accepted species thresholds. These genomic distinctions, together with differentiating physiological and chemotaxonomic traits, support the recognition of two novel species within the genus Aliikangiella. Accordingly, we propose Aliikangiella litoralis sp. nov. (type strain IMCC44359T) and Aliikangiella aequoris sp. nov. (type strain IMCC44632T; reference strain IMCC44653).
Description of Aliikangiella litoralis sp. nov.
Aliikangiella litoralis (li.to.ra′lis.; L. fem. adj. litoralis, coastal, of the shore)
Cells are Gram-stain-negative, strictly aerobic, motile rods, 0.3–0.5 µm in width and 1.7–2.7 µm in length. Colonies on marine agar are cream, circular, and convex. Growth occurs at 15–35℃ (optimum, 30℃), pH 6.0–9.0 (optimum, pH 8.0), and 1.0–4.0% (w/v) NaCl (optimum, 2.0%). Oxidase and catalase activities are positive. Hydrolyzes casein, Tween 20, and Tween 80, but not colloidal chitin, DNA, starch, and CM-cellulose. Esculin is not hydrolyzed. In API 20NE tests, nitrate reduction, and gelatinase are positive, whereas indole production, glucose fermentation, arginine dihydrolase, urease, and β-galactosidase activities are negative. Utilization of D-glucose, L-arabinose, D-mannose, D-mannitol, N-acetyl-glucosamine, D-maltose, potassium gluconate, capric acid, adipic acid, malic acid, trisodium citrate, and phenylacetic acid is negative. In API ZYM tests, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase, α-chymotrypsin, acid phosphatase, and naphthol-AS-BI-phosphohydrolase activities are positive, but lipase (C14), cystine arylamidase, trypsin, α-galactosidase, β-galactosidase, β-glucuronidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase activities are negative. In the GEN III MicroPlate, oxidizes L-histidine, L-serine, glucuronamide, L-malic acid, and acetoacetic acid. Predominant fatty acids are C14:0, C16:0, iso-C15:0, iso-C11:0 3-OH, and summed feature 9 (iso-C17:1 ω9c and/or C16:0 10-methyl). The major respiratory quinone is Q-8. Major polar lipids are phosphatidylethanolamine, phosphatidylglycerol, diphosphatidylglycerol, one unidentified aminolipid, and one unidentified lipid.
The type strain is IMCC44359T (= KCTC 18089T = JCM 37879T = HNIBRBA19635T) isolated from surface seawater off Jangbong Island, Republic of Korea. The genome size of the type strain is 5,951,045 bp and the DNA G + C content of 36.5%. The GenBank accession numbers for the 16S rRNA gene sequence and genome sequence are PX239437 and CP199292, respectively.
Description of Aliikangiella aequoris sp. nov.
Aliikangiella aequoris (ae'quo.ris.; L. gen. n. aequoris, of the sea)
Cells are Gram-stain-negative, strictly aerobic, motile rods, 0.4–0.8 µm in width and 1.4–3.8 µm in length. Colonies on marine agar are cream, circular, and convex. Growth occurs at 10–40℃ (optimum, 30℃), pH 6.0–9.0 (optimum, pH 7.0–8.0), and 0.5–7.5% (w/v) NaCl (optimum, 2.0–3.0%). Oxidase activity is positive and catalase activity is negative. Hydrolyzes casein, Tween 20, and Tween 80, but not colloidal chitin, DNA, starch, and CM-cellulose. Esculin is not hydrolyzed. In API 20NE tests, gelatinase is positive, whereas nitrate reduction, indole production, glucose fermentation, arginine dihydrolase, urease, and β-galactosidase are negative. Utilization of D-glucose, L-arabinose, D-mannose, D-mannitol, N-acetyl-glucosamine, D-maltose, potassium gluconate, capric acid, adipic acid, malic acid, trisodium citrate, and phenylacetic acid is negative. In API ZYM tests, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, and naphthol-AS-BI-phosphohydrolase activities are positive, but lipase (C14), trypsin, α-chymotrypsin, α-galactosidase, β-galactosidase, β-glucuronidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase activities are negative. Activities of valine arylamidase, cystine arylamidase, and acid phosphatase are variable. In the GEN III MicroPlate, oxidizes glucuronamide and L-malic acid. Oxidation of D-malic acid and bromo-succinic acid is variable. Predominant fatty acids are iso-C15:0 and summed feature 9 (iso-C17:1 ω9c and/or C16:0 10-methyl). The major respiratory quinone is Q-8. Major polar lipids are phosphatidylethanolamine, phosphatidylglycerol, diphosphatidylglycerol, one unidentified aminophospholipid, and three unidentified lipids. The genome size is approximately 3.75 Mbp and the DNA G + C content is 40.1–40.2%.
The type strain is IMCC44632T (= KCTC 18090T = JCM 37880T = HNIBRBA19636T), isolated from coastal seawater off Jangbong Island, Republic of Korea. The GenBank accession numbers for the 16S rRNA gene sequence and genome sequence are PX239438 and JBSKFV000000000, respectively.
Acknowledgments
This study was supported by the research grant “Survey of island-coastal indigenous organisms (Prokaryotes)” (HNIBR2026-A-2-07) from Honam National Institute of Biological Resources (HNIBR) and by the Mid-Career Research Program (NRF-2022R1A2C3008502) through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Korea.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information
The online version contains supplementary material available at https://doi.org/10.71150/jm.2602008
Table S1.
General genomic features of strains IMCC44359T, IMCC44632T, and IMCC44653 and the type strains of Aliikangiella species
Strains: 1, IMCC44359T; 2, IMCC44632T; 3, IMCC44653; 4, A. marina GYP-15T; 5, A. coralliicola M105T; 6, “A. maris” GXAS 306T.
jm-2602008-Supplementary-Table-S1.pdf
Table S2.
Carbon source oxidation patterns determined using the GENⅢ MicroPlate system for strains IMCC44359T, IMCC44632T, IMCC44653, A. marina KCTC 42667T, and A. coralliicola KCTC 72442T
Strains: 1, IMCC44359T; 2, IMCC44632T; 3, IMCC44653; 4, A. marina KCTC 42667T; 5, A. coralliicola KCTC 72442T. All data are from this study. +, positive; ‒, negative.
jm-2602008-Supplementary-Table-S2.pdf
Table S3.
Average amino acid identity (AAI; lower triangle) and percentage of conserved proteins (POCP; upper triangle) among strains IMCC44359T, IMCC44632T, IMCC44653, and related Aliikangiella species. Values represent percentages.
Strains: 1, IMCC44359T; 2, IMCC44632T; 3, IMCC44653; 4, A. marina GYP-15T; 5, A. coralliicola M105T; 6, A. maris GXAS 306T.
jm-2602008-Supplementary-Table-S3.pdf
Table S4.
KEGG module profiles of strains IMCC44359T, IMCC44632T, IMCC44653, Aliikangiella marina GYP-15T, Aliikangiella coralliicola M105T, and Aliikangiella maris GXAS 306T
jm-2602008-Supplementary-Table-S4.xlsx
Fig. S1.
Transmission electron micrographs of cells of strains IMCC44359T (A), IMCC44632T (B), and IMCC44653(C) after negative staining with 1% (w/v) uranyl acetate. Cells were grown on marine agar at 25°C for 5 days. Bar, 0.5 μm.
jm-2602008-Supplementary-Fig-S1.pdf
Fig. S2.
Two-dimensional thin-layer chromatogram of polar lipids of strains IMCC44359T (A), IMCC44632T (B), and IMCC44653 (C). PE, phosphatidylethanolamine; PG, phosphatidylglycerol; DPG, diphosphatidylglycerol; AL, unidentified aminolipid; APL, unidentified aminophospholipid; L, unidentified lipid.
jm-2602008-Supplementary-Fig-S2.pdf
Fig. 1.Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic placement of strains IMCC44359T, IMCC44632T, and IMCC44653. Bootstrap support values (≥ 70%) from the maximum-likelihood, neighbor-joining, and minimum-evolution methods (in that order) are shown at the nodes. Filled circles indicate nodes recovered by all three tree-building methods, whereas open circles denote nodes supported by two of the three methods. Scale bar, substitutions per nucleotide position.
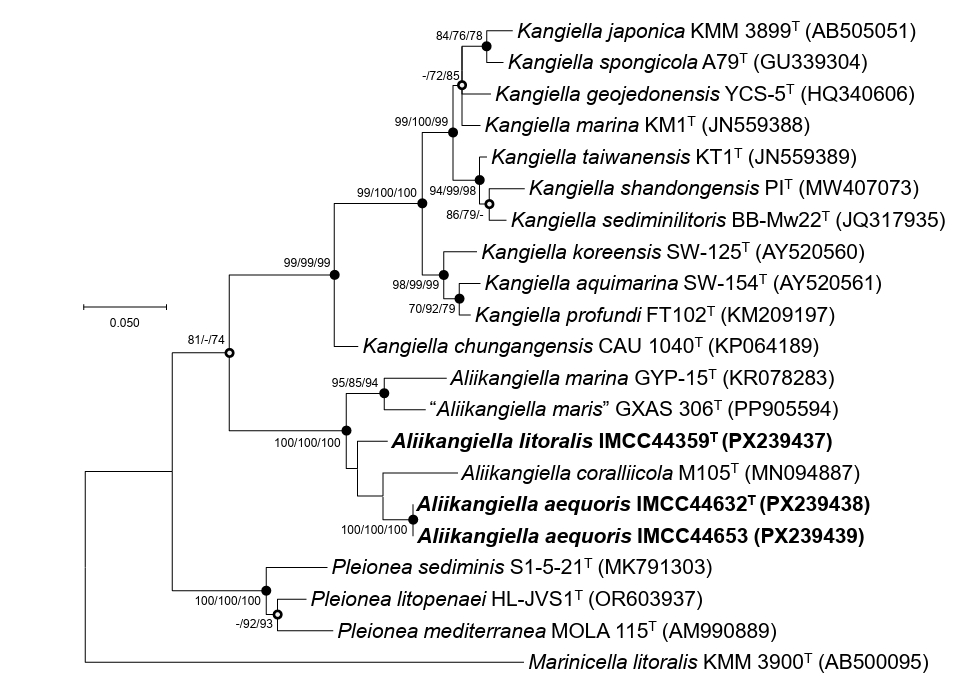
Fig. 2.Maximum-likelihood phylogenomic tree showing the relationships of strains IMCC44359T, IMCC44632T, and IMCC44653 to related type strains. The tree was reconstructed using GTDB-Tk (bac120 marker set) and RAxML based on concatenated amino acid sequences of 120 conserved single-copy marker genes. Bootstrap support values (≥ 70%) are shown at the nodes. Scale bar, substitutions per amino acid position.

Table 1.
Differential phenotypic characteristics of strains IMCC44359T, IMCC44632T, and IMCC44653 and the type strains of Aliikangiella species
Strains: 1, IMCC44359T; 2, IMCC44632T; 3, IMCC44653; 4, A. marina KCTC 42667T; 5, A. coralliicola KCTC 72442T: 6, “A. maris” GXAS 306T. Data are from this study unless otherwise indicated. All strains are positive for degradation of casein, Tween 20, and Tween 80, and enzyme activities for gelatinase, alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, and naphthol-AS-BI-phosphohydrolase. Detailed carbon source oxidation pattern is summarized in Table S2. +, Positive; ‒, negative; v, variable; nd, not determined.
|
Characteristics |
1 |
2 |
3 |
4 |
5 |
6c
|
|
Colony color |
cream |
cream |
cream |
pale yellow–green |
pale-yellow |
pale-yellow |
|
Growth at |
|
|
|
|
|
|
|
Temperature range (optimum, ºC) |
10–35 (30) |
10–40 (30) |
10–40 (30) |
15–37 (30) |
15–40 (25–30) |
15–37 (28–30) |
|
pH range (optimum) |
6–9 (8) |
6–9 (8) |
6–9 (7) |
6–9 (7–8) |
7–8 (7) |
5.5–10.5 (6–7) |
|
NaCl range (optimum, %) |
1–4 (2) |
0.5–7.5 (3) |
0.5–7.5 (2) |
1–5 (2–3) |
0–10 (1–3) |
0–4 (2–3) |
|
Oxidase |
+ |
+ |
+ |
+ |
– |
– |
|
Catalase |
+ |
– |
– |
+ |
+ |
+ |
|
Hydrolysis of: |
|
|
|
|
|
|
|
Starch |
– |
– |
– |
+ |
+ |
– |
|
Esculin |
– |
– |
– |
+a
|
+ |
nd |
|
API 20NE |
|
|
|
|
|
|
|
Nitrate reduction |
+ |
– |
– |
–a
|
+ |
nd |
|
β-Galactosidase |
– |
– |
– |
–a
|
+ |
nd |
|
API ZYM |
|
|
|
|
|
|
|
Lipase (C14) |
– |
– |
– |
+ |
–b
|
+ |
|
Valine arylamidase, Acid phosphatase |
+ |
– |
+ |
+ |
+b
|
v |
|
Cystine arylamidase |
– |
– |
+ |
+ |
+b
|
+ |
|
Trypsin |
– |
– |
– |
+ |
+b
|
+ |
|
α-Chymotrypsin |
+ |
– |
– |
+ |
+b
|
+ |
|
GEN III |
|
|
|
|
|
|
|
Gentiobiose, Sucrose, N-Acetyl-D-Glucosamine, N-Acetyl-β-D-Mannosamine, D-Fucose, L-Fucose |
– |
– |
– |
– |
+ |
nd |
|
L-Histidine, L-Serine |
+ |
– |
– |
– |
– |
nd |
|
D-Malic acid |
– |
+ |
– |
+ |
+ |
nd |
|
Bromo-succinic acid |
– |
+ |
– |
+ |
– |
nd |
|
Acetoacetic acid |
+ |
– |
– |
– |
+ |
nd |
Table 2.
Cellular fatty acid compositions (%) of strains IMCC44359T, IMCC44632T, and IMCC44653 and the type strains of validly published Aliikangiella species
Strains: 1, IMCC44359T; 2, IMCC44632T; 3, IMCC44653; 4, A. marina KCTC 42667T; 5, A. coralliicola KCTC 72442T; All data are from this study. All strains were cultured under identical conditions. Fatty acids representing < 1.0% of the total in all species were omitted. ‒, Not detected; Tr, traces (< 1.0%). Major fatty acids (> 10%) are shown in bold.
|
Fatty acid (%) |
1 |
2 |
3 |
4 |
5 |
|
Saturated |
|
|
|
|
|
|
C14:0
|
10.3 |
2.1 |
1.9 |
Tr |
1.0 |
|
C16:0
|
16.0 |
1.7 |
1.6 |
4.9 |
2.7 |
|
C18:0
|
Tr |
– |
– |
1.5 |
Tr |
|
Branched |
|
|
|
|
|
|
iso-C13:0
|
Tr |
1.2 |
1.1 |
Tr |
– |
|
iso-C14:0
|
Tr |
1.3 |
1.4 |
1.7 |
2.4 |
|
iso-C15:0
|
27.6 |
42.7 |
42.9 |
34.6 |
29.7 |
|
iso-C16:0
|
2.3 |
3.8 |
4.1 |
9.9 |
8.1 |
|
iso-C17:0
|
2.5 |
1.7 |
1.6 |
7.3 |
12.6 |
|
Hydroxy |
|
|
|
|
|
|
iso-C10:0 3-OH |
2.3 |
Tr |
– |
Tr |
Tr |
|
iso-C11:0 3-OH |
10.4 |
9.0 |
8.5 |
3.0 |
4.0 |
|
Unsaturated |
|
|
|
|
|
|
iso-F C15:1
|
Tr |
3.3 |
3.0 |
Tr |
1.3 |
|
C16:1 iso H |
– |
Tr |
Tr |
1.6 |
1.2 |
|
Summed Feature |
|
|
|
|
|
|
Summed feature 3*
|
5.1 |
5.4 |
5.1 |
5.3 |
9.6 |
|
Summed feature 8*
|
Tr |
– |
– |
– |
2.4 |
|
Summed feature 9*
|
19.2 |
26.9 |
27.4 |
25.5 |
20.1 |
References
- Bouras G, Page AJ, Watson M. 2024. Hybracter: Enabling scalable, automated, complete and accurate bacterial genome assemblies. Microb Genom. 10: 001244.ArticlePubMedPMC
- Chalita M, Kim YO, Park S, Oh HS, Cho JH, et al. 2024. EzBioCloud: A genome-driven database and platform for microbiome identification and discovery. Int J Syst Evol Microbiol. 74: 006421.ArticlePubMedPMC
- Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. 2022. GTDB-Tk v2: Memory friendly classification with the Genome Taxonomy Database. Bioinformatics. 38: 5315–5316. ArticlePubMedPMCPDF
- Chklovski A, Parks DH, Woodcroft BJ, Tyson GW. 2023. CheckM2: A rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods. 20: 1203–1212. ArticlePubMedPDF
- Collins MD, Jones D. 1981. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implications. Microbiol Rev. 45: 316–354. ArticlePubMedPMCLink
- Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, et al. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6: 1186–1199. ArticlePubMedPDF
- Felsenstein J. 1981. Evolutionary trees from DNA sequences: A maximum-likelihood approach. J Mol Evol. 17: 368–376. ArticlePubMedPDF
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 39: 783–791. ArticlePubMedLink
- Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, et al. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 309: 1242–1245. ArticlePubMed
- Han J, Lim Y, Kim M, Cho JC. 2025. Rubrivirga aquatilis sp. nov. and Rubrivirga halophila sp. nov., isolated from Korean coastal surface seawater. J Microbiol. 63: e2504017. ArticlePubMedPDF
- Hölzer M. 2024. POCP-nf: An automatic Nextflow pipeline for calculating the percentage of conserved proteins in bacterial taxonomy. Bioinformatics. 40: btae175.ArticlePubMedPMC
- Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 428: 726–731. ArticlePubMed
- Kim D, Park S, Chun J. 2021. Introducing EzAAI: A pipeline for high-throughput calculations of prokaryotic average amino acid identity. J Microbiol. 59: 476–480. ArticlePubMedPDF
- Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 16: 111–120. ArticlePubMedPDF
- Konstantinidis KT, Tiedje JM. 2007. Prokaryotic taxonomy and phylogeny in the genomic era: Advancements and challenges ahead. Curr Opin Microbiol. 10: 504–509. ArticlePubMed
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35: 1547–1549. ArticlePubMedPMC
- Li F, Xu MB, Pan LH, Li J, Lan CB, et al. 2025. Ammonifying and phosphorus-solubilizing function of Aliikangiella maris sp. nov. isolated from Phaeocystis globosa bloom and algal-bacterial interactions. Front Microbiol. 16: 1516993.ArticlePubMedPMC
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, et al. 2004. ARB: A software environment for sequence data. Nucleic Acids Res. 32: 1363–1371. ArticlePubMedPMC
- Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 14: 60.ArticlePubMedPMCPDF
- Minnikin DE, O’Donnell AG, Goodfellow M, Alderson G, Athalye M, et al. 1984. An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J Microbiol Methods. 2: 233–241. Article
- Oh S, Lim Y, Rajeev M, Cho JC. 2026. Robiginitalea rubriflava sp. nov. and Robiginitalea insularis sp. nov., isolated from coastal seawaters of the Yellow Sea. J Microbiol. 64: e2512009. ArticlePubMedPMC
- O’Leary NA, Cox E, Holmes JB, Anderson WR, Falk R, et al. 2024. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Sci Data. 11: 732.ArticlePubMedPDF
- Parte AC, Sardà Carbasse J, Meier-Kolthoff JP, Reimer LC, Göker M. 2020. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol. 70: 5607–5612. ArticlePubMedPMC
- Powers EM. 1995. Efficacy of the Ryu nonstaining KOH technique for rapidly determining gram reactions of food-borne and waterborne bacteria and yeasts. Appl Environ Microbiol. 61: 3756–3758. ArticlePubMedPMCLink
- Pruesse E, Peplies J, Glöckner FO. 2012. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 28: 1823–1829. ArticlePubMedPMCPDF
- Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, et al. 2014. A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol. 196: 2210–2215. ArticlePubMedPMCLink
- Richter M, Rosselló-Móra R, Glöckner FO, Peplies J. 2016. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 32: 929–931. ArticlePubMedPDF
- Riesco R, Trujillo ME. 2024. Update on the proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 74: 006300.ArticlePubMedPMC
- Rzhetsky A, Nei M. 1992. A simple method for estimating and testing minimum-evolution trees. Mol Biol Evol. 9: 945–967. Article
- Saitou N, Nei M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 4: 406–425. ArticlePubMed
- Sasser M. 1990. Identification of bacteria by gas chromatography of cellular fatty acids. MIDI technical note 101. MIDI Inc.. Link
- Seemann T. 2014. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 30: 2068–2069. ArticlePubMedPDF
- Stamatakis A. 2014. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30: 1312–1313. ArticlePubMedPMCPDF
- Tak H, Park MS, Cho H, Lim Y, Cho JC. 2024. Congregibacter variabilis sp. nov. and Congregibacter brevis sp. nov. within the OM60/NOR5 clade, isolated from seawater. J Microbiol. 62: 739–748. ArticlePubMedPDF
- Wang G, Tang M, Wu H, Dai S, Li T, et al. 2015. Aliikangiella marina gen. nov., sp. nov., a marine bacterium from the culture broth of Picochlorum sp. 122, and proposal of Kangiellaceae fam. nov. in the order Oceanospirillales. Int J Syst Evol Microbiol. 65: 4488–4494. ArticlePubMed
- Wang Y, Tang M, Wu H, Dai S, Li T, et al. 2020. Aliikangiella coralliicola sp. nov., a bacterium isolated from coral Porites lutea, and proposal of Pleioneaceae fam. nov. to accommodate Pleionea and Aliikangiella. Int J Syst Evol Microbiol. 70: 4278–4286. Article
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 1991. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173: 697–703. ArticlePubMedPMCLink
- Yang SH, Park MJ, Oh HM, Park DJ, Kwon KK. 2024. Flavivirga spongiicola sp. nov. and Flavivirga abyssicola sp. nov., isolated from marine environments. J Microbiol. 62: 11–19. ArticlePubMedPDF
- Yoon JH, Oh TK, Park YH. 2004. Kangiella koreensis gen. nov., sp. nov. and Kangiella aquimarina sp. nov., isolated from a tidal flat of the Yellow Sea in Korea. Int J Syst Evol Microbiol. 54: 1829–1835. ArticlePubMed
Citations
Citations to this article as recorded by



 ePub Link
ePub Link Cite this Article
Cite this Article